Review – DOI: 10.33594/000000858
Accepted 3.11.2025 - Published online 6.4.2026
Cellular Physiology and Biochemistry (60): 136 - 174
mTOR Signalling in Neurodegenerative Disorders: Unveiling Key Factors, Mechanistic Insights, and Possible Therapeutic Interventions
bDepartment of Biotechnology Engineering and Food Technology, Chandigarh University, Mohali 140413, India,
cDepartment of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak, Haryana, 124001, India,
dDepartment of Medical Laboratory Sciences, College of Applied Medical Sciences, University of Bisha, Bisha 61922, Saudi Arabia,
eCenter for Global Health Research, Saveetha Medical College, Saveetha Institute of Medical and Technical Sciences, India,
fDepartment of Biomedical Sciences, College of Health Sciences, Abu Dhabi University, P.O. Box 59911, Abu Dhabi, United Arab Emirates,
gSchool of Pharmaceutical Sciences, Lovely Professional University, Phagwara, Punjab 144411, India,
hSchool of Applied and Life Sciences, Uttaranchal University, Dehradun, 248007,
iDepartment of Biotechnology, College of Applied Sciences, University of Technology, Baghdad, Iraq,
jDepartment of Clinical pharmacology and Medicine, College of Medicine, Mustansiriyah University, Baghdad, Iraq,
kDepartment of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, Qassim University, Qassim, Saudi Arabia,
lDepartment of Biotechnology, Graphic Era Deemed to be University, Dehradun 248002, Uttrakhand,
mCentre for Research Impact and Outcome, Chitkara University, Punjab,
nSchool of Health Sciences and Technology, UPES, Dehradun, Uttarakhand 248007, India
Keywords
Abstract
Neurodegenerative diseases (NDDs) are defined by the gradual degeneration of neuronal cells, wherein the accumulation of misfolded proteins can lead to memory impairments, motor dysfunctions, and other deteriorations. Despite the widespread impact, there are currently no viable pharmaceuticals to treat these disorders. Currently, more than 30 million individuals globally endure severe health issues, with few therapeutic alternatives for their management. The mTOR protein is a crucial regulator of cell death signals, survival, growth, and the fate of neural cells. Targeted modulation of mTOR signaling holds the potential for mitigating neurodegeneration in Alzheimer's, Huntington's, Amyotrophic Lateral Sclerosis (ALS), Parkinson's, and other neurodegenerative conditions. However, the advancement of successful mTOR-targeted therapeutics necessitates careful consideration of multiple factors. These include addressing protein aggregation and comprehending the complex interactions of cell death pathways within the neurological system. Additionally, mTOR modulates diverse signaling pathways, encompassing growth factors and proteins, including PI3K (phosphoinositide 3-kinase), Akt, AMPK, and SIRT1. Understanding these connections is crucial for effective therapeutic development. By comprehending the intricacies of the mTOR pathway, researchers have the opportunity to uncover novel and more precise therapies, offering optimism for the future management of NDDs.
Introduction
The protein mTOR, also known as the mammalian target of rapamycin, acts as a cellular control center and significantly impacts aging, cellular senescence, and age-related diseases. It operates through two distinct complexes, mTOR complex 1 (mTORC1) and mTORC2, each with a unique composition and response to signals from other pathways, ultimately affecting cellular function [1, 2]. mTOR serves as the master regulator of protein synthesis, influenced by diverse factors such as brain-derived neurotrophic factor (BDNF), insulin-like growth factor (IGF)-1, vascular endothelial growth factor (VEGF), cytokines, and insulin, all of which act on kinase receptors and activate signal transduction pathways involving mTOR. mTORC1, a nutrient-sensitive protein complex inhibited by rapamycin [3], controls key growth regulators (e.g., cyclin D1, HIF-1α, c-myc) essential for cell growth and survival, and DNA damage from cellular stress [4]. Conversely, the role of mTORC2 is more complex and generally rapamycin-insensitive; its function depends on its interaction with the rapamycin-insensitive companion of mTOR (RICTOR) [5]. mTOR plays a crucial role in many fundamental cellular functions, including growth, survival, immune response, autophagy, apoptosis, and metabolism. However, mTOR dysfunction is linked to a wide range of diseases. This includes age-related impairment, arthritis, insulin resistance, various cancers, and problems with the nervous system. Disruptions in mTOR signaling are strongly implicated in the development of several neurodegenerative disorders (NDDs), including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington’s disease (HD), and Amyotrophic Lateral Sclerosis (ALS) [6]. AD is a progressive neurodegenerative disorder that primarily disrupts brain functions such as behavior, thinking, and memory, leading to a gradual decline in daily activities over time [1, 7,8]. The multifaceted nature of AD, with its numerous contributing factors, makes both prevention and treatment highly challenging. One of the key areas of investigation focuses on mTORC1, a cellular signaling pathway heavily influenced by various stimuli, including growth factors, insulin, amino acids, nutrients, energy levels, and cellular stresses and oxidative stress [9]. Moreover, scientific literature suggests that dysregulated mechanisms of cell survival and death may contribute to AD's development. For example, disruptions in the mTOR signaling pathways could also initiate events leading to AD pathogenesis [10]. PD, HD, and ALS are all neurodegenerative disorders with distinct causes and damage specific to different parts of the nervous system. PD is also just one of many NDDs that are projected to significantly impact the global population in the future. These progressive conditions lead to protein accumulation, PD (α-synuclein), HD (Htt), and ALS (TDP-43, SOD1), and impose a heavy burden globally [11–13]. However, they share common dysfunctional cellular processes, including the mTOR pathway, but no proper treatments are available to effectively cure these diseases. Additionally, many studies show that lowering mTORC1 activity can lengthen the lifespan in various organisms [14, 15]. Beyond its general functions, mTORC1 signaling is particularly interesting for its role in NDs. This is because mTORC1 not only controls protein building but also regulates their breakdown and the removal of cellular components through a process called autophagy. This cellular recycling system is crucial for preventing the accumulation of abnormal protein clumps, which contribute to diseases like AD, PD, ALS, and HD [16, 17]. As they share common dysfunctional cellular processes, including the mTOR pathway, a comprehensive approach targeting multiple aspects of the disease is crucial for effective management and for potentially mitigating the immense impact of treating NDDs. Studies on animals suggest that mTOR inhibitor drugs like rapamycin might have the ability to slow down the worsening of AD or even improve problems with thinking and memory that occur in AD patients [18–20]. However, according to data from clinicaltrials.gov, no clinical trials investigating the effects of rapamycin on delaying AD progression have been terminated to date [16]. Plants are emerging as a significant source of novel compounds that can modulate mTOR activity, offering potential therapeutic strategies for various diseases by harnessing the natural ability of plant-derived compounds to influence this crucial cellular pathway. The focus of this review is to comprehensively explore the multifaceted role of mTOR. It offers a thorough examination of mTOR complexes, their regulation, and their downstream impacts on cellular processes such as metabolism, autophagy, and aging, all of which play a critical role in AD, HD, PD, and ALS progression. Furthermore, the review critically analyzes the hurdles in translating these findings into effective clinical therapies for NDDs patients. It also delves into the critical involvement of diverse therapeutic agents in modulating mTOR activity that can hold therapeutic potential in ameliorating these debilitating conditions.
mTORC 1/2 complexes: Components, Functions, and Regulation
The mTOR is a ubiquitous 2549-amino acid serine/threonine protein kinase. It is mainly localized in the cytoplasm and belongs to the phosphatidylinositol 3-kinase-related kinase (PIKK) family. It is crucial for controlling fundamental cellular activities like survival, proliferation, apoptosis, and protein synthesis [21]. Its activity is positively correlated with phosphorylation at residues Thr2446, Ser2448, and Ser2481 in the catalytic domain. Notably, adjacent to this domain is the FKBP12–rapamycin-binding (FRB) domain, which serves as the specific binding site for the immunosuppressant rapamycin. Rapamycin forms a complex with FKBP12, which then binds to the FRB domain and disrupts the mTOR complex formation, inhibiting its activity [22]. The mTOR protein serves as the catalytic core of two distinct multi-protein complexes: mTORC1 and mTORC2 (Fig. 1 (A) and (B). These complexes are differentiated by their unique components, like upstream regulators, downstream targets, and sensitivity to rapamycin [23]. Both complexes possess shared subunits, including mTOR, mammalian lethal with DEP domain-containing mTOR-interacting protein (DEPTOR), SEC13 protein 8 (mLST8), and the Tti1/Tel2 complex. However, mTORC1 is distinctly characterized by the presence of the regulatory-associated protein of mTOR (Raptor) and the proline-rich Akt substrate of 40 kDa (PRAS40), whereas mTORC2 includes the rapamycin-insensitive companion of mTOR (Rictor), the mammalian stress-activated protein kinase-interacting protein 1 (mSin1), and the protein associated with Rictor 1/2 (Protor1/2) [24]. Raptor (150 kDa as a full-length scaffold protein) is an essential constituent of mTORC1 [25]. It plays a crucial role in complex formation, substrate recruitment, and recognition through its interaction with proteins containing the TOR signaling (TOS) motif, such as eukaryotic translation initiation factor 4E-binding proteins (4EBPs), p70 S6 kinase (S6K), and STAT3 (Hoeffer and Klann, 2010). Furthermore, Raptor functions as an amino acid-sensing molecule that regulates the subcellular localization and activation of mTORC1 [27]. Its ability to specifically compete with the rapamycin-FKBP12 complex for binding to FRB is a key mechanism in the regulation of mTORC1 activity. PRAS40 and DEPTOR serve as negative regulators of mTORC1 [28]. PRAS40 interacts with Raptor and inhibits mTORC1 by obstructing substrate access. mTORC1 activation leads to the phosphorylation and functional suppression of PRAS40 and DEPTOR, hence promoting downstream signaling [26]. mTORC2, distinguished by the presence of Rictor, typically exhibits resistance to acute rapamycin administration. Prolonged exposure to rapamycin can impair mTORC2 function, presumably by hindering the production of new Rictor proteins. This indicates that only preassembled mTORC2 complexes demonstrate resistance to rapamycin, likely due to steric hindrance that limits access to the FRB domain. Sin1, a component specific to mTORC2, is crucial for the stability of the complex, as its genetic ablation leads to embryonic mortality. mTORC2 functionally modulates cytoskeletal structure and phosphorylates essential kinases, including Akt. Akt activation indirectly enhances mTORC1 activity and inhibits FOXO transcription factors by obstructing their nuclear localization [29, 30]. The tuberous sclerosis complex (TSC), consisting of TSC1 and TSC2, serves as a vital negative regulator upstream of both mTOR complexes. TSC1/2 suppresses mTOR signaling through the small GTPase Rheb (Ras homolog abundant in brain) by enhancing its intrinsic GTPase activity, thereby converting active Rheb-GTP to inactive Rheb-GDP [31]. Numerous kinases modulate the TSC1/2 complex activity by phosphorylation, thus governing heterodimer formation. The result of these phosphorylation events—either activation or inhibition of mTOR—hinges on the particular amino acid residues that undergo phosphorylation. Among these kinases, glycogen synthase kinase 3β (GSK3β) phosphorylates TSC2, thereby facilitating the activation of the TSC1/2 complex and consequently inhibiting the mTORC1 activity [32].
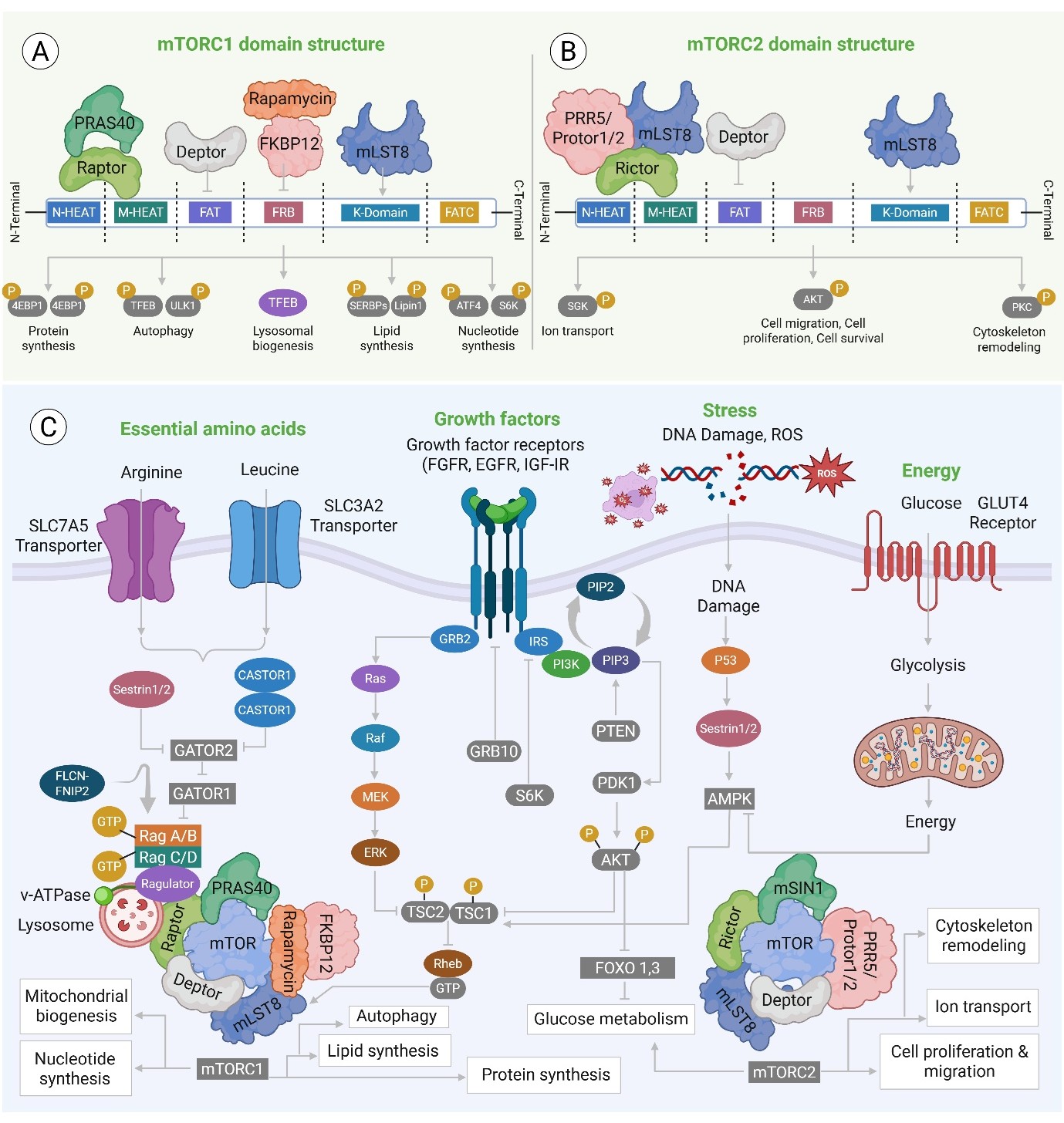
Fig. 1: (A) mTORC1: It acts as the master regulator of anabolism, promoting cell growth by activating effectors for Protein synthesis (S6K, 4EBP1), Lipid synthesis, and suppressing catabolic processes like Autophagy (via ULK1 inhibition). Overactivation of mTORC1 is a central driver of Tumor Proliferation and is implicated in Metabolic Syndrome and Neurodegenerative Disorders (due to impaired autophagy). (B) mTORC2 domain structure: It is distinguished by the rapamycin-insensitive subunit Rictor and mSIN1. mTORC2 phosphorylates AKT (at Ser473) to promote cell survival and regulates the cytoskeleton via PKC and ion transport via SGK. (C) The major upstream regulators of the mTOR complex: the mTOR network integrates signals from four major physiological inputs to control cell fate via Essential amino acids (Arginine, Leucine) activate mTORC1 via the Rag GTPases and GATOR complexes at the lysosome. Additionally, signaling via receptors (FGFR, EGFR) activates the PI3K-AKT pathway, which inhibits the tumor suppressors TSC1/TSC2 (mutated in Tuberous Sclerosis Complex), leading to mTORC1 activation. Low cellular energy (high AMP: ATP ratio) activates AMPK, which inhibits mTORC1 to conserve energy. Further cellular stress activates tumor suppressors like p53 and Sestrin,
mTOR Signaling: A Key Regulator of Cellular Fate
The mTOR is a serine/threonine kinase that is part of the PIKK family. The C-terminal region of this protein shares substantial structural similarity with the catalytic domains of both phosphatidylinositol 3-kinase (PI3K) and phosphatidylinositol 4-kinase (PI4K) [29]. mTOR serves as the catalytic core of mTORC1 and mTORC2 complexes, which are essential for regulating the intracellular signaling cascades critical for cellular homeostasis. The mTOR is vital in cell survival and proliferation by controlling a range of cellular processes, such as autophagy, protein synthesis, cell growth, and metabolism [33, 34]. Dysregulation, particularly overactivation, of mTOR signaling can lead to uncontrolled proliferation of cells and contribute to cancer [35, 36]. Beyond its role in cancer, mTOR also regulates immune cell differentiation and function, and it can inhibit apoptosis in tumor cells, aiding immune evasion. Consequently, mTOR serves as a crucial coordinator of cellular energy metabolism, proliferation, autophagy, and apoptosis. The mTOR protein is characterized by multiple conserved domains. Adjacent to the N-terminus, there is a sequence of HEAT repeats (huntingtin, elongation factor 3, protein phosphatase 2A, and TOR1), which are important for promoting the formation of a large surface area with a hydrophobic character that facilitates protein-protein interaction and membrane localization. The FAT domain (FRAP–ATM–TRRAP), spanning about 568 residues, interacts with the FATC domain at the C-terminus, thus revealing the kinase domain. This domain is vital for ATP binding and catalysis, and carrying out its catalytic function, and is therefore a target for ATP-competitive inhibitors [35, 36]. Rapamycin exerts its inhibitory effect on mTOR through a specific mechanism initially by forming a complex with FK506-binding protein 12 (FKBP12). Then this complex activates the FRB domain of mTOR, which induces a conformational change to inhibit kinase function. A negative regulatory domain (NRD) is also present, located between the KIN and FATC domains [37]. The mTOR signaling pathway is a crucial part of cellular communication networks, and the PI3K/AKT/mTOR cascade is a central hub in cellular communication. As shown in Fig. 1 (C), mTORC1 activity is governed by extracellular signals such as growth factors, hormones, and cytokines. PI3K phosphorylates PIP2 to form PIP3, which subsequently leads to the activation of AKT. The TSC1–TSC2 complex serves as a critical regulator that tightly controls the activity of AKT, and its inhibition can be alleviated by the activation of AMPK or REDD1 and the inhibition of Rheb. In addition, cellular stressors (e.g., energy deprivation, hypoxia, and genotoxic stress) can suppress mTORC1 activity via the activation of REDD1 or AMPK [37]. Amino acids are critical for activation of mTORC1, inducing the Rag GTPase heterodimer associations (RagA/B and RagC/D), which recruit the recruitment of mTORC1 to the lysosomal membrane, leading to its activation. Once stimulated, mTORC1 phosphorylates downstream targets like eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and ribosomal protein S6 kinase 1 (S6K1), favoring protein and lipid biosynthesis. mTORC1 also controls autophagy and the ubiquitin-proteasome system. In contrast, mTORC2 modulates cell metabolism, survival, and cytoskeleton organization by controlling its downstream targets AKT, protein kinase C (PKC), and serum/glucocorticoid-regulated kinase 1 (SGK1) [37, 38] Dysregulated mTOR signaling, particularly its overactivation, is a recognized contributor to the pathogenesis of a wide range of diseases. This includes its frequent observation in various types of cancer [39] and is associated with neurological diseases such as AD, PD, and HD [40–42]. In addition, mTOR signaling is also involved in metabolic disorders, including diabetes, obesity, and aging abnormalities [43]. Therefore, pharmacological targeting of mTOR signaling has provided an avenue for therapeutic intervention by mitigating aberrant growth factor signaling, delaying disease progression, and potentially offering significant advantages in the treatment of NDs.
Driving Cellular Fate: The Molecular Mechanisms of mTOR Activation
mTOR, as a fundamental cellular regulator, is controlled by three major upstream signals: nutrient availability, environmental stressors, and immune system responses [44]. These diverse signals are integrated to activate mTOR, a process involving multiple signaling pathways ultimately. One crucial pathway involves Ras homolog enriched in brain (RHEB), a small GTPase protein found on the lysosome. RHEB functions depend on its association with guanine nucleotide. When GTP is bound to RHEB, it can engage with mTORC1, a protein complex, and bring it to the lysosome. This interaction is necessary for the direct activation of mTORC1 [45, 46]. Separately, the PI3K/Akt signaling pathway serves as the primary mechanism by which growth factors stimulate mTORC1. This occurs when growth factors attach to receptor tyrosine kinases (RTK), these receptors activate PI3K, an enzyme that produces a specific lipid known as PIP3. PIP3 formation leads to the activation of PDK1 and Akt, two proteins engaged in the signaling cascade. Akt then phosphorylates and inhibits TSC2, a core element of the TSC complex [47, 48]. Since the TSC complex serves as a GTPase-activating protein, it facilitates the inactivation of RHEB. Akt's inhibition of TSC2 indirectly activates mTORC1 by allowing RHEB to become more active and can promote mTORC1 activation [49]. Another pathway downstream of RTKs, which can facilitate mTORC1 activation, is the Ras/Erk/RSK1. This pathway suppresses the TSC complex and also indirectly activates mTORC1. Moreover, RSK1 can directly activate mTORC1 by phosphorylating Raptor, a protein within the mTORC1 complex [50–52]. The PI3K/AKT signaling is the key pathway by which antigens, cytokines, and other immune signals induce mTORC1 activation [53, 54]. For example, TLR signaling is known to activate PI3K/AKT to activate mTORC1, which acts to regulate the inflammatory response [55, 56]. As cellular metabolism critically regulates mTORC1, nutrient and energy availability strongly influence its activity. AMPK, a protein that regulates energy, gets activated by a reduction in glucose levels and an elevation in the AMP/ATP ratio. AMPK indirectly suppresses the mTORC1 through the TSC phosphorylation, thus augmenting its capacity to decrease the mTORC1 activity [57]. Gwinn et al. found that AMP-activated protein kinase (AMPK) directly suppresses mTORC1 through Raptor phosphorylation [58]. Moreover, mTORC1 activity is stringently controlled by amino acid supply via the RAG-GTPases and Ragulator [59]. RAG A/B and RAG C/D constitute a heterodimer that associates with the lysosome, facilitated by Ragulator. The heterodimer of GTP-bound RAG A/B and GDP-bound RAG C/D drives mTORC1 translocation to the lysosome, leading to its activation [27]. GATOR1 inhibits mTORC1 activation by suppressing RAG A/B. GATOR2 mitigates this inhibition. Leucine, arginine, and glutamine are notably proficient in activating mTORC1 among amino acids [60]. Cytoplasmic leucine levels are detected by Sestrin2 (SESN2), which reduces its affinity for GATOR2 upon leucine binding. This enables GATOR2 to engage with GATOR1, hence diminishing its inhibitory influence on RAG and mTOR [61–63]. CASTOR1, akin to SESN2, identifies cytoplasmic arginine and suppresses GATOR2 under conditions of arginine scarcity. Abundant arginine alleviates CASTOR1's suppression of GATOR2, resulting in mTOR activation [64]. Lysosomal arginine further activates mTORC1 by stimulating the RAG-Ragulator complex through the amino acid transporter SLC38A9. Crucially, vacuolar H+-ATPase (v-ATPase) detects lysosomal amino acids and is essential for RAG-Ragulator-mediated mTORC1 activation [65, 66]. Adenosine diphosphate ribosylation factor-1 (Arf1), a small GTPase, stimulates mTORC1 in a manner that is independent of RAG in response to glutamine. Glutamine is transformed into α-ketoglutarate (αKG), which activates RAG by modifying its nucleotide state, ultimately resulting in mTORC1 activation [67, 68]. The mechanisms underlying mTORC2 activation remain incompletely elucidated; nonetheless, it is established that growth factor-induced-RTK activation activates mTORC2 via PI3K [69]. PIP3 interacts with mSin1 (the regulatory subunit of mTORC2), thereby diminishing its inhibitory effect. PI3K facilitates the connection between mTORC2 and the ribosome, leading to the activation of mTORC2. Furthermore, glucose deprivation-induced AMPK activation phosphorylates mTOR, thereby directly activating mTORC2 [38, 70].
mTOR and the Autophagy Nexus: Gatekeeper of Cellular Homeostasis
mTOR, through its complex mTORC1, plays a vital role in regulating cellular functions such as translation and autophagy. It acts by phosphorylating various substrates, including 4EBP, Ulk1, p70S6K, and Atg13. Autophagy is a crucial cellular process for metabolic equilibrium and survival [71]. This process involves the formation of a double-membraned structure autophagosome, that engulfs and transports cytoplasmic material to the vacuole for degradation [72]. Dysregulation of autophagy is linked to various human pathologies, such as lysosomal storage disorders and neurological conditions [73, 74]. The process is generally regulated by several autophagy-related (Atg) proteins and progresses through three distinct phases: initiation, elongation, and maturation. The initiation phase commences with the establishment of a phagophore formation at the phagophore assembly site. The elongation phase entails the phagophore expansion, whereas the maturation phase concludes with autophagosome-lysosome fusion to form an autophagolysosome. mTORC1 is a key regulator of autophagy, which under nutrient-replete-conditions inhibits autophagy by suppressing and phosphorylating Ulk1, Atg13, and AMBRA1, thereby blocking the development of the phagophore [75, 76]. Conversely, when nutrients are scarce or when rapamycin is present, mTORC1 activity diminishes, resulting in the dephosphorylation and activation of Ulk1. This activated Ulk1 then triggers the autophagic cascade by phosphorylating Atg13, FIP200, and as well and itself [77]. The Ulk1/Atg13/FIP200/Atg101 protein complex is fundamental for autophagy, since it promotes the formation of autophagosomes, specialized structures that sequester damaged organelles or proteins for destruction. Its primary role is to recruit other crucial autophagy proteins and initiate autophagosome formation. The activation of this complex initiates a series of processes, leading to the activation of another critical complex, including the class III PI3 kinase Beclin-1 and Vps34. Vps34, an enzyme that phosphorylates lipids, is crucial for the de novo synthesis of autophagosomes, and its enzymatic activity is substantially enhanced upon binding to Beclin-1 [78]. A crucial phase in autophagy involves the conjugation of LC3, a microtubule-associated protein, with phosphatidylethanolamine (PtdEtn). This process involves the initial cleavage of LC3 by Atg4 to form LC3-I, followed by its conjugation to PtdEtn via Atg7 and Atg3, leading to the production of LC3-II [79, 80]. LC3-II, a crucial autophagy protein, is affixed to both surfaces of the autophagosome membrane. During fusion with lysosomes, LC3-II on the cytosolic face is converted back to LC3-I by Atg4. In contrast, LC3-II on the inner (luminal) surface undergoes degradation. Autophagosomes are then actively transported to lysosomes along microtubules, facilitated by a motor protein dynein. The subsequent merging of these two organelles creates an autolysosome, where the sequestered cellular components are broken down by lysosomal enzymes. mTORC1 exhibits a dual regulatory role in autophagy; it not only impedes autophagy but also negatively impacts lysosome formation, which is indispensable for cellular degradation [79]. Emerging evidence suggests that mTORC1 regulates lysosome formation via TFEB, a transcription factor that governs genes associated with lysosomal function [81]. In conditions of nutrient deprivation, mTORC1 inhibition facilitates TFEB's nuclear translocation, hence enhancing both the generation of autophagosomes and their subsequent fusion with lysosomes. This ultimately improves autophagy to counteract nutritional deficits [81, 82].
mTOR at the junction: Modulating Apoptosis via Autophagy
Apoptosis, a form of programmed cell death characterized by a series of events involving caspase activation. These proteolytic enzymes are triggered in response to stimuli like mitochondrial damage or TNF ligand-receptor binding [83]. mTOR modulates apoptosis through its complex relationship with autophagy, although the precise underlying mechanisms are not yet fully understood. Autophagy itself plays a dual role in regulating apoptosis, while mTOR affects this process via intricate autophagy-dependent pathways. Notably, the PI3K/AKT/mTOR signaling pathway suppresses apoptosis by promoting autophagy. However, a study by Han et al. showed that oxidative stress caused by hydrogen peroxide can initiate apoptosis; however, this effect can be mitigated by the PI3K/AKT/mTOR pathway, which inhibits autophagy [84]. Furthermore, studies show that when mTOR is inactive, it promotes apoptosis via autophagy activation, as evidenced by the fact that autophagy inhibitors can reduce apoptosis induced by mTOR inhibitors [85, 86]. The complex interplay between autophagy and apoptosis enables mTOR to have a dual influence on apoptosis. mTOR acts as a suppressor of autophagy; when this suppression occurs during cellular stress, it can trigger apoptosis. However, when autophagy is active, it functions as a protective mechanism that helps the cell avoid apoptosis. For instance, in a murine model of sepsis, mTOR inhibition attenuates autophagy, consequently triggering death in CD4+ T cells [87]. Recent studies also highlight the AKT/mTOR pathway's significance in modulating autophagy and apoptosis [88, 89]. This pathway can suppress autophagy, resulting in enhanced apoptosis. The mechanisms by which autophagy have been extensively investigated. One key mechanism involves mTOR inhibition, which triggers autophagy and promotes the degradation of the anti-apoptotic protein Bcl-2, thereby triggering apoptosis [90]. Additionally, certain autophagy proteins, including ATG5 and ATG12, might directly facilitate apoptosis either through interactions with death-related proteins or by inactivating anti-apoptotic proteins such as Bcl-2 [91]. Conversely, autophagy can also suppress apoptosis by engulfing and degrading caspase-8, a key apoptotic enzyme. The interaction of ATG7 with caspase-9 obstructs its translocation to the autophagosome, further influencing the apoptotic cascade [92].
Significance of mTOR in Brain Function
The mTOR is highly expressed in the brain, predominantly in neurons and also in glial cells. During embryonic development, mTOR signaling is essential for enhancing neuronal survival and proliferation in response to growth hormones and guidance cues, such as insulin and insulin-like growth factor 1 (IGF-1). The coordinated function of mTORC1 and mTORC2 is crucial for appropriate neuronal and cerebral development, despite the underlying mechanisms being only partially elucidated [93]. mTOR signaling promotes neurite outgrowth, encompassing the elongation of dendrites and axons [94] (Fig. 2). In dendritic morphogenesis, mTOR hyperactivation promotes dendritic branching and elevates the quantity of immature filopodia-like protrusions, while diminishing the density of mature dendritic spines [95]. The activation of mTORC1 facilitates protein and lipid biosynthesis, hence enhancing cellular growth and plasma membrane expansion, which are essential for axon guidance in neurodevelopment. Additionally, local mTORC1-dependent protein—and maybe lipid—synthesis facilitates the structural maturation of axons and dendrites. mTORC2, recognized for its modulation of actin cytoskeletal dynamics, presumably enhances growth cone motility and is crucial to neurite pathfinding and elongation [32]. As the brain develops, the mTOR signaling pathways that control neuronal division and migration during development become crucial regulators of synaptic plasticity and cognitive functioning in the mature brain [96]. In post-mitotic neurons, mTOR regulates various processes, including synaptic plasticity, neuronal polarity, neurotransmission, proteostasis, metabolic regulation, and cellular stress responses, including DNA repair [97]. In the adult central nervous system, mTOR activity is essential for multiple forms of synaptic plasticity, especially long-term potentiation (LTP) in the hippocampus, which facilitates learning and memory via protein synthesis-dependent synaptic enhancement [98]. Moreover, the mTOR pathway facilitates synaptic plasticity by meticulously regulating the time and positioning of new protein production. Research indicates that mTOR facilitates gene expression regulated by BDNF expression, highlighting numerous critical proteins associated with synaptic plasticity, learning, and memory [32, 95, 99], the production of synaptic proteins, including calcium/calmodulin-dependent protein kinase II alpha (CaMKIIα) and postsynaptic density protein 95 (PSD-95), is also dependent upon mTOR signaling [100, 101]. In the aging brain, mTOR is crucial for maintaining proteostasis by balancing the equilibrium between protein synthesis and autophagy, thus averting the buildup of toxic protein aggregates that may lead to neurodegeneration [97]. Beyond its localized functions in neurons, mTOR also governs systemic physiological processes. Within the hypothalamus, it acts as an energy sensor to regulate food consumption and sustain overall energy homeostasis [102]. Additionally, mTOR plays a significant role in modulating the hypothalamic-pituitary-gonadal axis, affecting the onset of puberty, responding to external light stimuli, and adjusting circadian rhythms through its effect on neurons in the suprachiasmatic nucleus [99, 103].
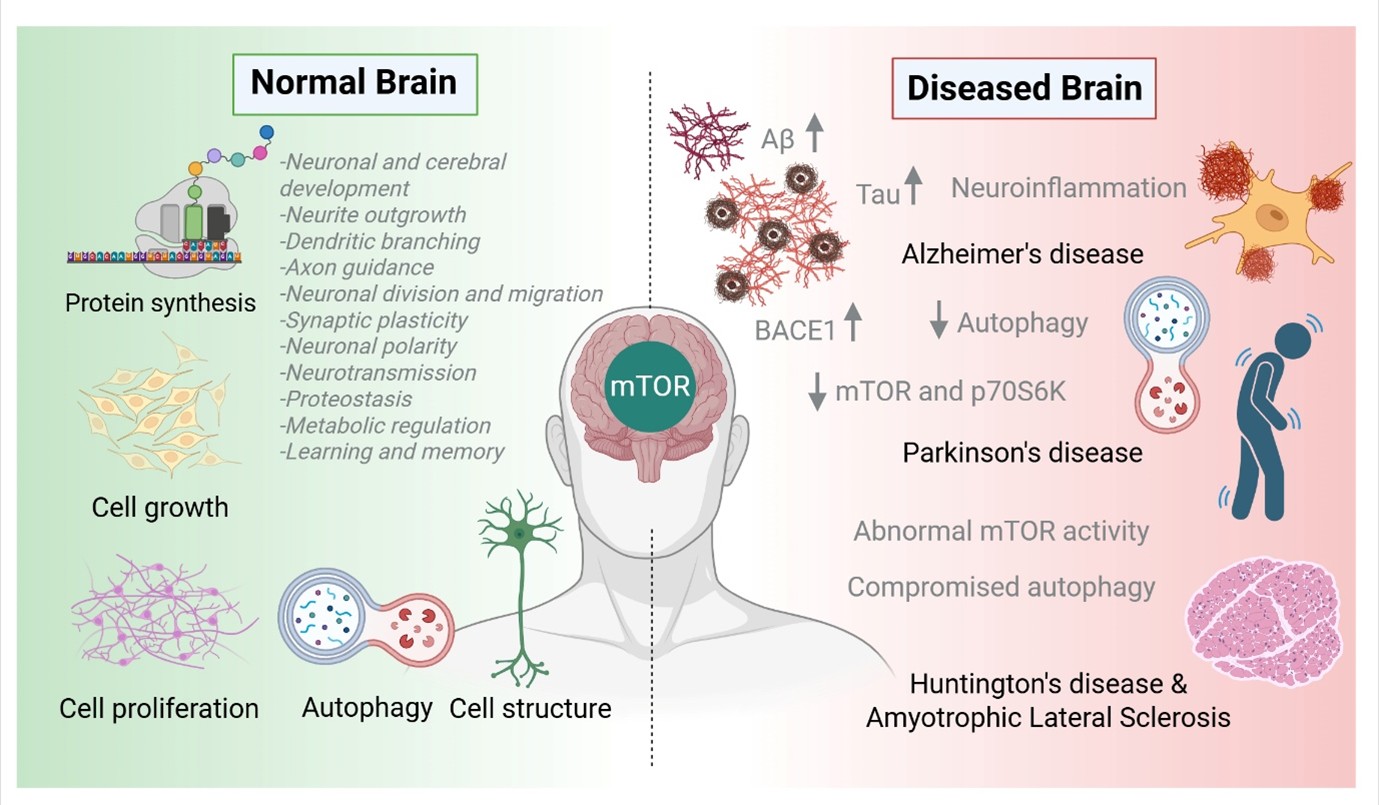
Fig. 2: Clinical significance of mTOR in normal vs diseased brain: In neurodegenerative disorders like Alzheimer's, Parkinson's, Huntington's disease, and ALS, abnormal mTOR activity and compromised autophagy are linked to pathological features like increased Aβ and Tau levels, BACE1 reduction, and neuroinflammation.
mTOR and neuroinflammation
The mTOR signaling pathway is a critical intracellular regulator that profoundly affects immune responses, especially inside the central nervous system (CNS). mTOR significantly impacts the activation and function of microglia, the resident immune cells of the CNS, thereby promoting neuroinflammation. Extensive research has demonstrated that mTOR signaling regulates T cell development and function, escalating inflammatory responses. Both external stimuli and internal dysregulation can activate the nervous system's immune responses, hence engaging the mTOR pathway and promoting neuroinflammatory processes. Activation of mTOR signaling can lead to heightened activation and secretion of glial cells, which are crucial for immunological defence and metabolic regulation, leading to neuroinflammation. Furthermore, the mTOR signaling pathway and its activity are associated with the regulation of cell membrane integrity. Under stress, damage, or infection in nerve cells, mTOR activation may exacerbate membrane breakdown and ionic imbalance, hence facilitating neuroinflammatory reactions. Neuroinflammation, the inflammatory breakdown of brain tissue caused by infection, autoimmune responses, or other injuries, is a defining feature of the onset and advancement of NDDs [104]. Chronic neuroinflammatory signaling can lead to progressive neuronal damage and death, hence increasing vulnerability to conditions such as AD, PD, and multiple sclerosis. Early neuroinflammation is especially harmful, as it can provoke synaptic instability, glial dysfunction, and neuronal demise, all accelerating disease progression. This process involves the release of inflammatory mediators, including cytokines, chemokines, oxidative stress markers, and other neurotoxic agents, which collectively exacerbate neurodegeneration [105]. Dysfunctional immune cells and inflammatory mediators are central to disease onset and progression in many NDDs, including epilepsy [106]. The mTOR signaling pathway critically regulates neuroinflammatory responses, particularly through the modulation of microglial activation. Microglia, the CNS's innate immune cells, become activated in response to infection, injury, or inflammatory stimuli, subsequently releasing various cytokines, including pro-inflammatory chemokines and interleukins [107, 108]. While this activation recruits other immune cells and aids tissue repair, excessive microglial activation may lead to sustained inflammation, worsening the pathophysiology of disorders such as AD and multiple sclerosis [109]. Activated microglia generate many cytotoxic agents, including ROS, prostaglandin E2 (PGE2), proteolytic enzymes, nitric oxide (NO), and pro-inflammatory cytokines, which may exacerbate neuronal damage [110]. The mTOR signaling pathway facilitates macrophage activation by regulating the innate immune system. Inhibition of mTOR has been linked to reduced microglial viability and a reduction in pro-inflammatory responses. Specifically, mTOR activity boosts the survival of EOC2 microglial cells and increases NO synthase 2 (NOS2) expression under hypoxic conditions in BV2 microglia, hence promoting a pro-inflammatory phenotype. Conversely, mTOR inhibition reduces NOS2 expression and NO production in response to cytokines, and also lowers microglial proliferation and intracellular cyclooxygenase levels, especially when combined with cytokine therapy or mTOR inhibitors such as RAD2 [111]. mTOR signaling is critical for T cell development and function, while also influencing neuroinflammation. mTOR dictates the fate of CD4⁺ T cells by promoting their differentiation into effector subsets and modifying their functional responses. CD4⁺ Th1 cells typically become anergic (inactive) upon antigen recognition without co-stimulation [112]. However, mTOR activity can circumvent this anergic state by acting as a metabolic co-stimulatory signal, therefore promoting T cell activation [113]. Inhibition of mTOR impedes this activation, intensifying T cell anergy and disrupting essential metabolic pathways vital for T cell activity [114]. Furthermore, mTOR inhibition enhances the development and activity of regulatory T cells (Tregs), which are crucial for immune balance and preventing excessive inflammation [115]. Studies indicate that mTOR inhibitors have shown the potential to reduce cytokine-dependent T cell proliferation while promoting Treg-mediated immunological tolerance [111]. Furthermore, mTOR activation reduces the expression of lymph node-homing receptors, such as CD62L and CCR7, hence facilitating the T cell migration towards inflamed tissues [113]. These findings underscore mTOR signaling's pivotal role in regulating immunological responses and neuroinflammatory processes, establishing it as a crucial factor in the development of NDs and a potential therapeutic target.
mTOR signaling in Neurodegenerative disorders
Alzheimer’s disease
Alzheimer’s disease AD is a progressive, age-associated NDDs marked by a gradual and permanent deterioration of cognitive functions, including memory, learning, and executive abilities [105, 116]. Pathologically, AD is primarily distinguished by two hallmark features, the extracellular deposits of β-amyloid (Aβ) peptides forming amyloid plaques and intracellular aggregates of hyperphosphorylated tau protein resulting in neurofibrillary tangles (NFTs) [117]. These features signify complex disruptions in neuronal function and integrity, ultimately leading to the observed symptoms in affected individuals. AD is the most widespread NDDs worldwide, presently impacting over 20 million individuals. The number is anticipated to increase significantly, with an estimated 135 million individuals forecast to be diagnosed by 2050. Familial AD, characterized by an autosomal dominant inheritance pattern, constitutes less than 2% of all cases and is predominantly linked to mutations in the genes producing amyloid precursor protein (APP) and presenilin-1 and -2 (PSEN1, PSEN2), which are essential for Aβ formation. The vast majority of AD cases are sporadic and complex in nature. Age is the predominant risk factor, closely followed by the presence of the apolipoprotein E4 (APOE ε4) allele [118]. The other contributing factors encompass metabolic illnesses like diabetes and obesity, cerebrovascular anomalies, brain injuries, and diverse hereditary predispositions [119]. The neuropathological examinations of AD brain uncover a variety of molecular changes in addition to plaques and tangles. These encompass oxidative stress, disturbances in energy metabolism, mitochondrial dysfunction, proteostasis impairment through compromised autophagy and proteasome activity, degradation of the blood-brain barrier, excitotoxicity resulting from glutamate dysregulation, diminished acetylcholine signaling, and persistent inflammation [120]. These alterations are coordinated by or associated with dysregulations in critical intracellular signaling networks such as intracellular calcium, c-Jun N-terminal kinases, PKR, nicotinic acetylcholine receptor signaling, AMPK, and silent mating-type information regulator 2 homolog 1 (Sirt1), and PI3K/Akt/ mTOR. The abnormal activation of the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway has become a key pathogenic characteristic in AD [121] (Fig. 2). Recent studies have revealed an indirect association between the mTOR signaling pathway and Aβ pathology via its upstream regulator, Ras homolog enriched in brain (Rheb), which is observed to be downregulated in the brains of AD patients. The decrease in Rheb levels is associated with heightened expression of β-site APP-cleaving enzyme 1 (BACE1), the rate-limiting enzyme that facilitates Aβ synthesis in neurons [122, 123]. Both in vitro and in vivo studies have shown that Rheb modulates BACE1 degradation through proteasomal and autophagic pathways, independent of mTOR, by directly interacting with BACE1 [122]. Tau hyperphosphorylation, another key characteristic of AD, has been linked to elevated mTOR activity and its downstream signaling pathways [124]. In Drosophila tauopathy models, tau hyperphosphorylation amplifies mTOR signaling, leading to neurodegeneration—a process that can be reversed through genetic or pharmacological mTOR inhibition [125, 126]. Furthermore, mTOR activation exacerbates tau pathology by boosting its localization and secretion, suppressing protein phosphatase 2A (PP2A), and augmenting tau mRNA translation through p70S6K activation [127, 128]. The antidiabetic medication biguanide metformin inhibits mTOR, hence preventing tau hyperphosphorylation in both in vitro and in vivo settings [129]. Investigations of postmortem human AD brain tissues have revealed hyperactivation of the PI3K-Akt-mTOR signaling pathway. This elevation encompasses downstream effectors, including p70 ribosomal S6 kinase (p70S6K), 4E-BP1, and eukaryotic elongation factor 2, all of which influence modified protein production and cellular metabolism [124, 130–132]. Notably, only mTOR complex 1 (mTORC1), and not mTORC2, seems to be activated in the hippocampus during the advanced stages of AD [131]. This hyperactivation is especially significant in people with mild cognitive impairment (MCI) and clinical AD, but not in individuals with preclinical AD, indicating that mTOR dysregulation is a progressive phenomenon in AD pathogenesis [133]. AD, another characteristic is cerebral insulin resistance, seen as reduced glucose uptake and metabolism in neurons. This metabolic abnormality is associated with paradoxical hyperactivation of the insulin receptor (IR)-PI3K-Akt signaling cascade. Evidence suggests that soluble Aβ oligomers can attach to insulin receptors, promoting their internalization and removing IR from neuronal dendrites, thus replicating the insulin resistance phenotype observed in AD brains [134]. The degree of mTOR activation in animal models of AD has produced contradictory results, dependent on the particular transgenic mouse model and age. Prior investigations indicated diminished mTOR activity and lowered levels of its downstream effector, p70S6K, in two APP/PS1 transgenic mouse lines, specifically in the hippocampus at 6 months and in the cortex at 12 months of age [135]. Conversely, other researchers have noted hyperactivation of the mTOR pathway in 3xTg-AD animals and in the hippocampus of non-transgenic mice administered Aβ oligomers [136]. The hyperactivation of mTOR produced by Aβ may be associated with the phosphorylation of PRAS40, a positive modulator of mTOR activity [136–138]. Comparable variability has been observed in in vitro experiments. Utilizing Aβ1-42, a decrease in mTOR activation and p70S6K expression in both mixed primary cultures and neuroblastoma cells was observed [135, 139]. A similar suppression of mTOR signaling was noted in peripheral blood mononuclear cells (PBMCs) from AD patients, seemingly mediated through the PKR-p53-TSC1/TSC2-REDD1 pathway [140, 141]. In contrast, several investigations indicated heightened mTOR activation following Aβ25-35 administration or the expression of the familial AD mutation (7PA2) in CHO cells [136, 142, 143]. The combined in vivo and in vitro evidence indicates that Aβ influences mTOR signaling in a dose-dependent manner: lower, more physiological Aβ levels appear to augment mTOR activity, whereas higher Aβ concentrations inhibit it [144]. This duality underpins a neural dichotomy, wherein mTOR suppression correlates with apoptotic neurons, but mTOR hyperactivation, accompanied by increased Akt and p70S6K activity, is related to the development of NFTs [130, 145]. Neuroinflammation, while not a defining characteristic, significantly contributes to the progression of AD [105]. Microglia, the brain's intrinsic immune cells, demonstrate diminished efficacy in clearing Aβ as AD progresses, similar to macrophages [146]. T lymphocytes have been noted in proximity to Aβ plaques and activated microglia [147, 148]. The mTOR pathway governs the differentiation of T-helper cell subsets and the generation of cytokines [149]. mTORC1 specifically inhibits pro-inflammatory cytokines, including IL-12, IL-23, IL-6, and TNF-α, by suppressing NF-κB activity, while promoting anti-inflammatory cytokines such as IL-10, TGF-β, and type I interferons [150, 151]. Further research demonstrates reduced lymphocyte responsiveness to the mTOR inhibitor rapamycin in AD patients relative to healthy controls, signifying a more extensive disruption of immunological signaling [152]. mTOR is pivotal in modulating autophagy, a cellular breakdown mechanism that is compromised in AD [153]. Both human AD brains and transgenic mouse models demonstrate the increase of autophagic vesicles (AVs) in dystrophic neurites, potentially worsening disease pathology by increasing Aβ generation within AVs [154–157]. Various mechanisms that regulate autophagy—such as mTOR, AMP-activated protein kinase (AMPK), glycogen synthase kinase-3β (GSK3β), and calcium signaling are impaired in AD [158–160]. Moreover, reduced expression of beclin-1, a critical initiator of autophagy, coupled with inadequate lysosomal acidification, leads to compromised proteolysis and buildup of autophagic vacuoles [161, 162]. The efforts to reinstate autophagy in AD have had inconsistent outcomes. The re-expression of beclin-1 and administration of rapamycin in transgenic AD mice have decreased amyloid accumulation and tau protein levels [163, 164]. In contrast, the induction of autophagy by rapamycin in APP mutant cell lines has been linked to elevated Aβ synthesis. Interventions that boost lysosomal acidification, such as GSK3β inhibition (e.g., L803-mts) or the injection of nicotinamide (a NAD⁺ precursor), facilitate Aβ clearance and diminish tau pathology [165, 166]. The results demonstrate a stage-dependent relationship between autophagy and Aβ dynamics: initial autophagy activation may improve Aβ clearance, whereas later autophagic dysfunction may worsen pathogenesis [144]. These findings highlight the pivotal role of mTOR signaling dysregulation in AD, connecting critical clinical characteristics including Aβ accumulation, tau hyperphosphorylation, neuroinflammation, autophagy dysfunction, neuronal shrinkage, and cognitive decline. Consequently, targeting mTOR and its related pathways signifies a possible treatment strategy for altering the progression of AD.
Parkinson’s disease
Parkinson’s disease is a progressive NDDs that impacts roughly 2% of adults over 60 years of age, rendering it the second most prevalent neurodegenerative condition following AD. Clinically, PD is defined by motor symptoms including resting tremor, stiffness, bradykinesia, and postural instability. Alongside these motor problems, patients frequently encounter non-motor symptoms such as cognitive deficits, depression, and sleep disorders [167]. PD neuropathologically affects dopaminergic neurons, especially in the substantia nigra pars compacta and the striatum, as well as many non-dopaminergic systems, including the locus coeruleus, raphe nuclei, nucleus basalis of Meynert, hypothalamus, and pedunculopontine nucleus [168]. A pathological feature of PD is the aggregation of α-synuclein-positive intracytoplasmic inclusions referred to as Lewy bodies [169]. The exact origin of PD remains unclear; however, age is the key risk factor. There are various environmental factors, including pesticides (particularly paraquat and rotenone), head injuries, rural residency, use of well water, agricultural employment, and β-blocker usage, that have been associated with heightened vulnerability to the condition [170, 171]. Additionally, genetic variants of PD are present. Mutations in the SNCA gene, responsible for encoding α-synuclein, were the initial mutations associated with familial PD [172]. Further genes implicated in autosomal dominant PD comprise LRRK2, VPS35, eIF4G1, DNAJC13, and CHCHD2, whereas autosomal recessive variants typically presenting with early onset are connected with mutations in Parkin, PINK1, and DJ-1 [173–176]. Numerous cellular processes are impaired in PD, encompassing protein aggregation, intracellular trafficking, proteolytic systems (such as autophagy and the ubiquitin-proteasome system), mitochondrial function, and inflammatory responses [177–181]. A notable molecular characteristic in PD is the downregulation of the PI3K-Akt-mTOR pathway (Fig. 2). The postmortem examinations of PD brains demonstrate increased concentrations of REDD1, a powerful mTOR inhibitor, especially within dopaminergic neurons [182]. The upregulation may stem from the loss or mutation of Parkin, which is recognized for regulating REDD1 expression and contributing to neurodegeneration [183]. These findings are supported by data from experimental models of PD. In catecholaminergic PC12 cells exposed to 6-hydroxydopamine (6-OHDA), REDD1 expression is elevated and correlates with diminished Akt and mTOR activity [184]. Likewise, MPP⁺ (a neurotoxic byproduct of MPTP) diminishes phosphorylated mTOR and its downstream targets, namely p70S6K, eIF4E, and 4E-BP1, in neuroblastoma cells, hence impairing protein synthesis [185]. In PC12 cells, Rodríguez-Blanco et al. noted that MPP1 inhibited the Akt/mTOR pathway and the autophagic process due to the accumulation of reactive oxygen species [186]. Similarly, MPTP-treated mice in vivo demonstrate diminished mTOR and p70S6K activity in the striatum and frontal cortex, which correlates with cognitive impairments [187, 188]. Notably, although mTOR signaling is diminished in PD, prolonged L-DOPA administration the most efficacious treatment for motor symptoms can paradoxically stimulate striatal mTORC1 and provoke dyskinesia (L-DOPA-induced dyskinesia, or LID). This impact is achieved through extracellular signal-regulated kinases (ERKs), which inhibit TSC2, sustaining a sequential activation of Rheb and mTOR phosphorylation and activation of the mTORC1 component, Raptor [189]. The elimination of Rhes, a GTPase abundant in the striatum and an activator of mTOR, in 6-OHDA animal models diminishes LID and mTORC1 signaling, enhancing motor outcomes irrespective of ERK signaling. Dysfunction of autophagy is a significant characteristic of PD etiology [190]. Genetic mutations linked to familial PD, such as LRRK2, UCH-L1, PINK1, Parkin, and DJ-1, disrupt multiple facets of the autophagic process. LRRK2 mutations, for instance, impede chaperone-mediated autophagy and augment 4E-BP phosphorylation, hence elevating protein translation while diminishing oxidative stress tolerance [191, 192]. Mutations in PINK1 and Parkin impair mitophagy, resulting in mitochondrial malfunction and oxidative stress, while the precise function of DJ-1 is still ambiguous [193–195]. The A53T mutant variant of α-synuclein has been demonstrated to hinder autophagy while enhancing mTOR/p70S6K signaling, thereby facilitating neurodegeneration [196, 197]. The autophagy inhibition, exemplified by Bafilomycin A1, blocks the degradation of α-synuclein, promoting its aggregation into deleterious oligomers. These oligomers then worsen neuroinflammation and accelerate neuronal degeneration [179, 198, 199]. Hence, these findings highlight the critical significance of mTOR and autophagy dysregulation in the development and progression of PD.
Huntington’s disease
HD is an autosomal dominant neurological condition characterized by an expanded polyglutamine (CAG) repeat within the HTT gene situated on chromosome 4 [200]. While in healthy individuals, CAG repeat counts between 11 to 36, patients with HD often display expansions of 42 or more. Notably, the longest expansions (exceeding 66 repeats) are frequently associated with juvenile-onset forms of the disease. This specific genetic mutation results in the synthesis of mutant huntingtin protein (mHtt), characterized by an abnormally elongated polyglutamine tract at its N-terminus, leading to a harmful gain-of-function that drives disease progression [201]. HD is marked by a gradual deterioration of motor control. Patients initially exhibit choreic, involuntary movements that resemble a dance-like hyperkinetic condition [202]. This eventually evolves into a hypokinetic phase characterized by stiffness and bradykinesia [203] Cognitive symptoms frequently manifest years before clinical diagnosis and advance concurrently with motor symptoms [204]. Deficiencies in learning, attention, planning, visuospatial skills, and emotional perception are frequently documented. The illness generally initiates between the ages of 30 and 40 and advances over a span of 10 to 20 years, ultimately resulting in mortality[202]. Nonetheless, HD individuals may exhibit significant variations in age of onset and clinical symptoms, despite comparable CAG repeat lengths, indicating the potential role of genetic modifiers such as PPARGC1A (PGC-1α) and NMDA receptor genes (GRIN2A, GRIN2B), which may affect the age of onset [205, 206]. HD is characterized neuropathologically by the loss of efferent medium spiny neurons of the striatum, especially caudate and putamen, accompanied by extensive cortical atrophy [207, 208]. A distinguishing feature of HD, unlike other NDs, is the significant decrease (up to 50%) in BDNF, essential for neuronal viability [209]. Besides mHtt aggregates, HD pathophysiology encompasses mitochondrial malfunction, proteostasis failure, vesicular trafficking abnormalities, oxidative stress, and excitotoxicity [178, 202]. The mTOR signaling pathway, crucial for cell proliferation and autophagy regulation, is impaired in HD (Fig. 2). In the initial, presymptomatic stages, mTOR is seemingly overactivated; but, throughout symptomatic phases, particularly in postmortem HD brains, it is hypoactive and sequestered within mHtt aggregates [210]. This sequester is associated with reduced activity of downstream mTOR targets, including p70S6K and 4E-BP1, leading to compromised mRNA translation [211]. Significantly, normal Htt can augment mTOR activity in amino acid-deficient settings through its interaction with Rheb, a GTPase that activates mTOR. Mutant Htt has increased affinity for Rheb, potentially resulting in aberrant mTOR activation in the early stages of the disease [212]. The experimental research in HD mouse models has shown that the deletion of TSC1, negative regulator of mTORC1 in the striatum, expedited disease progression, whereas Rheb overexpression reinstated mTORC1 activity, augmented autophagy, promoted motor function, and safeguarded striatal neurons. Notably, while Rheb levels are consistent in HD patients, the striatal GTPase Rhes has comparable therapeutic effects upon overexpression. Rhes additionally amplifies mHtt toxicity by functioning as a SUMO E3 ligase, facilitating mHtt sumoylation [210]. In addition to mTOR-dependent pathways, Rhes can modulate autophagy independently of mTOR. In vitro, Rhes interferes with Bcl-2's inhibitory association with Beclin-1, hence initiating autophagy. This function is inhibited by Htt (both normal and mutant), which competes with Bcl-2 for Rhes binding [213]. Moreover, normal Htt directly activates ULK1, a crucial initiator of autophagy, by displacing mTORC1 from ULK1, so facilitating the assembly of the ULK1-Atg13-FIP200 complex essential for autophagosome formation [214]. Htt also serves a scaffolding function in the p62-mediated identification of ubiquitinated cargo. Conversely, polyQ-expanded mHtt seems to undermine these autophagic functions. The removal of the polyQ tract in Htt significantly improves autophagy and extends lifespan in murine models, indicating that polyQ growth disrupts Htt's typical role in selective autophagy [215].
Amyotrophic lateral sclerosis
ALS is a rapidly advancing, lethal neurodegenerative condition characterized by the degeneration of motor neurons in the central nervous system that govern voluntary muscular movements. Clinical signs encompass muscular weakness, stiffness, paralysis, and, in certain instances, cognitive deficits [216]. ALS generally manifests between the ages of 35 and 50, with the majority of patients experiencing respiratory failure within 3 to 5 years; however, approximately 10% may survive for a decade or longer. Approximately 90–95% of ALS cases are sporadic, devoid of familial history or identifiable risk factors [217]. Conversely, familial genetic form of ALS is frequently associated with mutations in the superoxide dismutase 1 (SOD1) gene, which encodes an antioxidant enzyme responsible for detoxifying free radicals [218]. These mutations result in a deleterious gain-of-function, causing protein misfolding, aggregation, and cellular distress [219]. Mutant SOD1 can be excreted, stimulate microgliosis, and induce apoptosis [220]. Additional genetic variants linked to ALS encompass alterations in the TDP-43 and FUS genes [221, 222]. The pathogenesis of the disease likely entails mitochondrial malfunction, immunological dysregulation, and impaired protein clearance [223, 224]. In SOD1G93A transgenic mice, a prevalent model for ALS, motor symptoms manifest approximately at 15 weeks, coinciding with a reduction in mTOR pathway activity and its downstream PI3K-Akt-p70S6K signaling inside the spinal cord (Fig. 2). Concurrently, autophagic markers, including p62 and LC3-II, increase, signifying compromised autophagy [225]. Notably, hyperexcitability and heightened p70S6K activation are evident in the cortical neurons of presymptomatic animals at one month, despite unchanged mTOR levels [226]. It is suggested that mutant SOD1 aggregates may sequester mTOR, so compromising its neuroprotective functions, a mechanism similarly observed in HD [227]. These findings underscore the detrimental significance of mutant protein aggregation and impaired mTOR signaling in the pathogenesis of ALS.
Therapeutic modulation of mTOR and its signaling networks in neurodegenerative disorders
In the last few decades, significant advancements have been achieved in understanding the intricate mTOR signaling network in the mammalian brain. The dysregulation of mTOR activity has become increasingly linked to aging and the etiology of AD and other NDDs (Table 1). Scientists have discovered several ways to influence mTORC1 activity and autophagy. Some compounds, like theophylline, target proteins upstream of mTORC1. Others, like rapamycin, directly interact with mTORC1 itself. There are even approaches that don't involve drugs, like caloric restriction. This dietary practice seems to activate natural molecules in the body (like AMPK) that then regulate mTORC1 [228]. Researchers have also identified numerous new molecules, including rapalogs (such as temsirolimus, everolimus, and sirolimus), mTORC modulators, DL001, NV-5440, and NR1 [229, 230]. These compounds are under investigation for their therapeutic potential in treating and managing NDs by altering mTOR and its associated signaling pathways through diverse mechanisms. The constraints of existing treatments for NDs, including adverse effects and disease advancement, have catalyzed investigations into alternative medicines. Medicinal plants have significant promise because of their varied cellular and molecular effects. These herbs can augment antioxidant defences, diminish pro-inflammatory cytokines, and mitigate inflammation, all of which contribute to neuroprotection. Their capacity to influence several signaling pathways further substantiates their preventative potential. NDs arise from a multifaceted interaction of elements, encompassing environmental impacts, oxidative stress, protein depletion, inflammation, and predominantly, aging. The plant-derived natural chemicals present a significant avenue for investigating novel therapies for these severe illnesses [231, 232]. The natural substances such as Pongamol, Baicalein, β-asarone, and Kaempferol (refer to Supplemental Table 1), utilized in traditional medicine, are being examined for their potential efficacy in treating NDs by regulating mTOR and its associated pathways [41, 233–235]. Moreover, researchers are investigating compounds that regulate mTOR and autophagy activity, a crucial cellular mechanism. Additional intriguing domains encompass apelins, myokines, and pharmacological agents that target PPARα, PTEN, and mGluR5. Therefore, research is needed to understand exactly how each compound works and the specific pathways involved in regulating autophagy. However, the understanding of the precise links between mTOR signaling and NDDs remains limited. Prolonged activation or inhibition of mTOR signaling could have significant consequences, underscoring the need for further research. Despite comprehensive preclinical studies suggesting therapeutic potential, clinical application is constrained, with few innovative medicines demonstrating success in human trials.
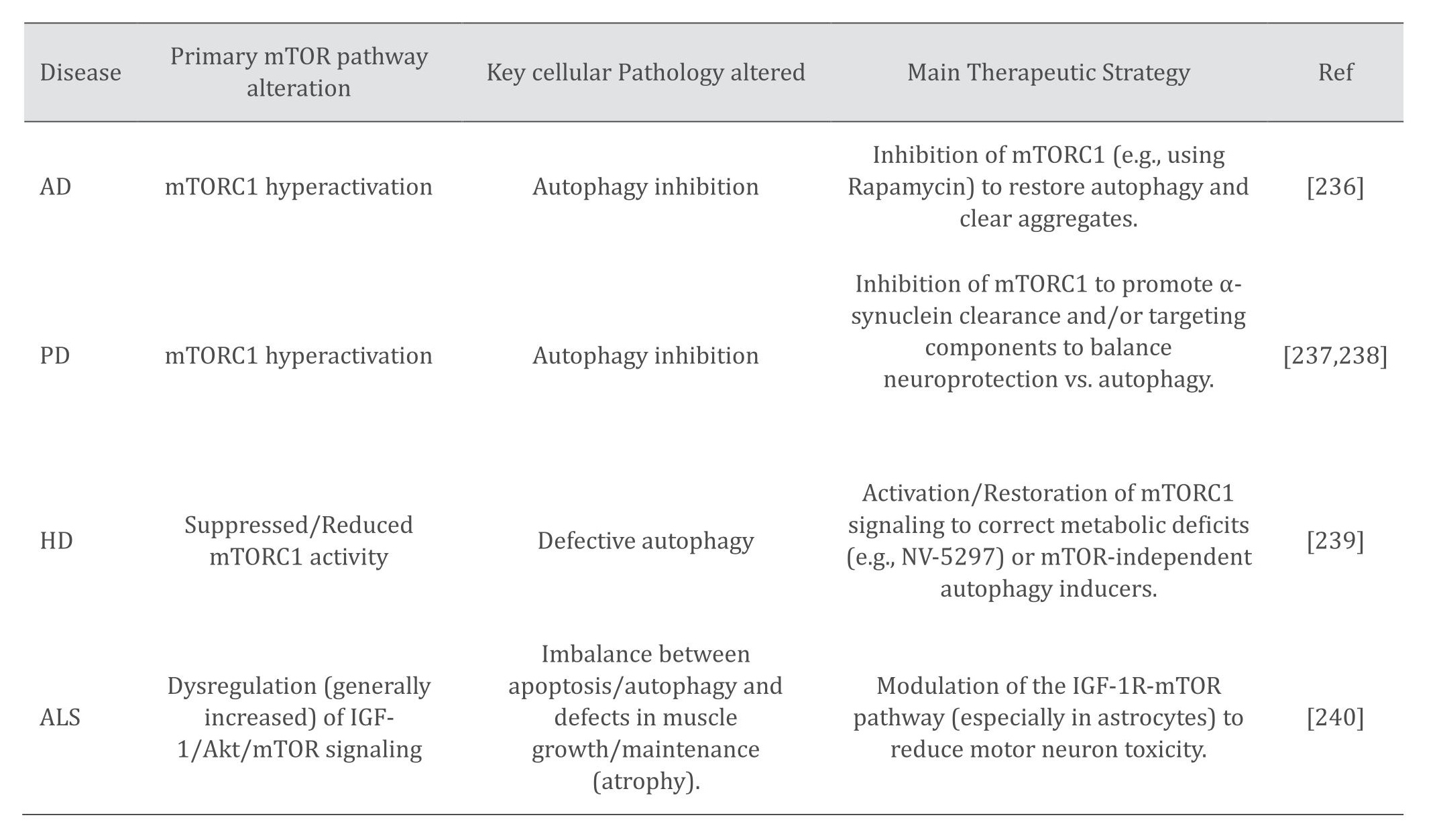
Table 1: mTOR Pathway Alterations and Therapeutic Targets in Major Neurodegenerative Diseases
mTOR modulation in Alzheimer’s disease
Rapamycin, a potent mTOR inhibitor, has garnered considerable interest due to its lifespan-extending and anti-aging properties, even in non-AD populations. The benefits are primarily due to its inhibition of mTORC1, which enhances autophagy and reduces overall protein production, thereby promoting cellular homeostasis. Research using animals has consistently shown that rapamycin and its analogs can improve cognitive impairments in AD models, indicating possible disease-modifying effects. The effects of rapamycin on synaptic and mitochondrial function seem to be influenced by apolipoprotein E (APOE) genotype. In asymptomatic E4FAD animal models, rapamycin augmented glutamate-glutamine cycling, ameliorated mitochondrial function, and promoted tricarboxylic acid cycle activity. In contrast, rapamycin enhanced inhibitory neurotransmission, non-neuronal activity and glycolysis in E3FAD animals. The genotype-dependent variations advocate that rapamycin may curtail the risk of AD in cognitively intact APOE4 carriers by ameliorating brain metabolic processes [40]. Besides neuronal metabolism, mTOR inhibition has shown efficacy in alleviating memory deficits and reinstating neurovascular coupling (NVC) in AD animal models [236]. Cerebrovascular change, also a hallmark of AD, involves inadequate neurovascular coupling, crucial for blood flow modulation to activate the brain regions. Rapamycin administration reinstates neurovascular coupling by reactivating endothelial nitric oxide synthase (eNOS) activity and mitigating mTOR-mediated suppression of neuronal and non-neuronal nitric oxide synthase pathways. Such observations underscore the profound significance of mTOR as a pivotal element in cognitive decline and NVC impairment in AD [236]. mTOR signaling also regulates blood-brain barrier integrity, another crucial factor impacted in AD, which is likewise regulated by mTOR signaling. Research has shown that rapamycin, by increasing tight junction protein expression and reducing matrix metalloproteinase-9 (MMP-9) activity, maintains BBB integrity and homeostasis, hence underscoring its therapeutic potential in neurovascular health [241]. Recently, Magnolol, a natural product, has been identified as a neuroprotective agent that activates the AMPK/mTOR/ULK1 signaling cascade, suppressing apoptosis and facilitating autophagy. In APP/PS1 mice, it regulated the pro- and anti-apoptotic markers (e.g., Bax, cleaved caspase-9, Bcl-2), reduced Aβ accumulation, improved cognitive function, while also elevating the expression of autophagy markers such as Beclin-1 and LC3-II [242]. Pongamol similarly produces pro-autophagic and anti-inflammatory actions by modulating the Akt/mTOR pathway. Pongamol diminished the production of pro-inflammatory cytokines (IL-1β, TNF-α, COX-2, iNOS), decreased tau phosphorylation and Aβ accumulation, and promoted autophagic flow through the overexpression of Beclin-1 and LC3-II in LPS-induced BV2 microglial cells and AD mice models [233]. Additionally, Traditional Chinese medicine formulations have shown effectiveness in modulating mTOR signaling in AD. Further, Danggui Shaoyao San (DSS), together with its constituent components Suangan (SG) and Xingan (XG), improved cognitive impairments and neuroinflammation in Aβ1-42-injected mice through the activation of AMPK/mTOR-mediated autophagy. DSS treatment elevated postsynaptic density protein 95 (PSD-95), reduced APP and phosphorylated tau, and influenced autophagy-related proteins (LC3, Beclin1, p-AMPK, p-mTOR, and p62), with DSS demonstrating enhanced therapeutic efficacy [243]. Other research assessed drugs like tadalafil and bergapten in a sporadic AD model produced by streptozotocin. These medicines enhanced cognitive function, diminished Aβ and tau pathology, and regulated a network of signaling pathways, including PI3K/Akt, GSK-3β, mTOR, Wnt/β-catenin, and cGMP/PKG. Their effects were additionally associated with elevated BDNF expression and reduced neuroinflammation [244]. The geniposide, extracted from gardenia fruit, has demonstrated neuroprotective effects through the modulation of the mTOR signaling cascade. Its treatment in APP/PS1 mice resulted in enhanced autophagic markers (Beclin1, LC3-II), increased cognitive performance, decreased Aβ accumulation, and altered phosphorylation patterns of mTOR, Akt, and 4E-BP1 [111]. Further, Tricetin (TRN), a flavonoid from honey and wheat, has demonstrated anti-inflammatory and neuroprotective properties in AD models. TRN improved memory, restored autophagy, and reduced toxic Aβ and tau buildup by targeting the PI3K/Akt/mTOR signaling pathway, indicating its potential as a therapeutic drug [245]. Recently, the traditional Chinese formulation Guben-Jiannao Ye (GBJNY) has exhibited its therapeutic efficacy in enhancing cognition and circadian rhythms in AD model mice. GBJNY decreased Aβ accumulation, reinstated circadian gene expression, and adjusted PI3K/Akt/mTOR signaling, consequently supporting its role in homeostatic regulation and neuroprotection [246]. Moreover, Glycoprotein NMB (GPNMB), which facilitates autophagy through the mTOR inhibition, has also displayed significant neuroprotective attributes. In APP/PS1 mice, GPNMB overexpression was observed to reduce Aβ levels and improve neurobehavioral outcomes. The autophagic benefits were abolished by 3-MA, hence validating mTOR-dependent autophagy as the principal mechanism [247]. Notably, non-pharmacological methods like exposure to 40 Hz gamma frequency stimulation also influence mTOR-related pathways. In vitro studies indicated reduced Aβ release, tau phosphorylation, and mTOR activity through the mTOR/4E-BP1/Tau pathway, implying therapeutic potential of rhythmic brain stimulation [276]. The mTOR signaling pathway in microglia is fundamentally associated with Trem2 expression and lysosomal biogenesis. In 5XFAD animals, the deletion of Tsc1 in microglia resulted in mTOR activation, increased expression of Trem2, and improved Aβ clearance, but simultaneous removal of Trem2 abolished these results. This identifies Trem2 as a downstream effector of mTOR in the microglial-mediated clearance of amyloid [277]. Furthermore, the activation of the TFEB, a principal regulator of the autophagy-lysosome pathway, has been recognized as an mTOR-dependent approach to alleviate AD-like pathology, especially in diabetic encephalopathy. Augmenting TFEB expression facilitated autophagic clearance and diminished neuronal apoptosis. The STAT2-SIRT4-mTOR pathway has emerged as a regulatory mechanism in AD. Increased SIRT4 levels in AD lead to neuronal death and Aβ accumulation, while the inhibition of SIRT4 through STAT2 modulation reverses memory impairment and diminishes Alzheimer's pathogenesis via mTOR signaling [278]. In the context of Down syndrome (DS), abnormal hyperactivation of the PI3K/Akt/mTOR pathway has been associated with the early emergence of AD-like symptoms. Targeting this axis may postpone disease advancement and enhance neurological results in Down syndrome-associated AD [279]. A notable target is Forkhead box G1 (FoxG1), which facilitates autophagy and suppresses Aβ-induced neuroinflammation through the AMPK/mTOR pathway. FoxG1 promotes neuronal survival, inhibits NLRP3 inflammasome activation, and boosts cognitive performance in AD models, highlighting its potential as a therapeutic option [280]. The mTOR signaling pathway also plays an important role in modulating autophagy, neuroinflammation, cerebrovascular integrity, and synaptic function in AD. In addition, although the direct inhibition of mTOR is difficult to achieve, indirect regulation of upstream regulators of mTOR, such as GPCRs, provides a potentially achievable approach. Due to their ‘drugable’ properties, GPCRs may offer a targeted intervention into the cross-talking PI3K, MAPK, and mTOR signaling network and thereby expand the therapeutic options for AD and related NDs [281]. In recent studies, the therapeutic value of mesenchymal stem cell (MSC)-derived exosomes (MSC-exos) on AD has been investigated. In an AlCl₃-induced AD rat model, the interventions comprised MSC-exosomes with or without autophagy regulators. The findings indicated that MSC-exosomes were able to improve memory, alleviate Aβ deposits and phosphorylation of tau, and enable neurogenesis and synaptic function. They suppressed astrogliosis and neuroinflammation by modulating microRNAs and autophagy, the PI3K-Akt-mTOR pathway. Histological and molecular studies confirmed these observations, indicating that MSC-exosomes are a promising therapeutic strategy for AD [282].
mTOR modulation in Parkinson’s disease
PD is characterized by the gradual loss of dopamine-producing neurons, buildup of α-synuclein, and dysfunctional autophagy. A growing body of evidence emphasizes the essential role of the mTOR signaling pathway in the regulation of autophagy, neuronal survival, and mitochondrial function. This suggests that modulating mTOR signaling through various natural products, drugs, transcription factors, and microRNAs may be potential neuroprotective strategies for PD intervention. Numerous phytochemicals and bioactive substances exhibit neuroprotective potential by modulation of mTOR-related pathways. For instance, Cordycepin (nucleoside analog) demonstrated neuroprotective properties in PD by reducing neuroinflammation, preserving dopaminergic neurons, and improving motor function. Moreover, Proteomic analysis attributes these benefits through suppressed inflammation, the enhancement of autophagy, and the modulation of the PI3K/AKT/mTOR and ERK/JNK pathways [248]. Piperine (PIP), the primary alkaloid in Piper longum, also provides neuroprotection by augmenting autophagy mediated by the gut-brain axis. In 6-OHDA-induced PD mice, PIP safeguarded dopamine neurons and reduced α-synuclein buildup. It also ameliorated motor and gastrointestinal dysfunction by suppressing the PI3K/AKT/mTOR pathway and facilitating α-synuclein clearance in both the substantia nigra and colon [237]. Schisandra Decoction (Sch D), which has been employed in PD management by focusing on the PI3K/AKT/mTOR pathway. In MPTP-induced PD mice, Sch D reduced α-synuclein buildup, suppressed autophagy, and improved motor function. Its neuroprotective effects may arise from the inhibition of oxidative stress and dysfunctional autophagy, necessitating additional investigation into its antioxidant characteristics [249]. Baicalein also demonstrates neuroprotection by diminishing cell death and increasing dopamine levels. It also helps in reinstating mitochondrial homeostasis and integrity maintenance. It functions by restoring SIRT1 expression and inhibiting miR-30b-5p, which in turn activates the AMPK/mTOR pathway to induce mitophagy and promote neuronal survival [234]. In a similar study, oral administration of Baicalin over four weeks markedly improved cognition, reduced neuronal apoptosis, and modulated the expression of essential signaling proteins, including mTOR, AKT, and GSK-3β. The results indicated a correlation between reduced α-synuclein levels and improved motor function, suggesting Baicalin's therapeutic efficacy via modulation of the mTOR/AKT/GSK-3β pathway [252]. Kaempferol (KAE), a flavonoid, offers neuroprotection through the regulation of autophagic flux. It has been shown to reduce α-synuclein accumulation, safeguard dopaminergic neurons, and improve motor function. Additionally, it stimulates autophagy via the overexpression of LC3 and Beclin-1, alongside mitochondrial restoration and mTOR inhibition [235]. Morus alba, Sanggenol L (SL), another flavonoid, was examined for its therapeutic efficacy against rotenone-induced injury in SK-N-SH neuroblastoma cells. It decreased ROS, oxidative stress, and apoptosis by modulating apoptotic markers and the PI3K/AKT/mTOR signaling pathway [251]. Interestingly, Cilostazol, primarily known as a vasodilator, has recently been found to alleviate rotenone-induced PD pathology. Its therapeutic benefits arise from its multi-target modulation, including modifying PI3K/Akt/mTOR signaling, blocking the HMGB1/TLR4 inflammatory cascade, and activating the Nrf2/HO-1 antioxidant pathway [250]. Curcumin, a polyphenolic substance, mitigates the pathogenic A53T α-synuclein mutation by inhibiting mTOR/p70S6K signaling and restoring autophagy. Thus, promoting α-synuclein clearance suggests curcumin's potential in both hereditary and idiopathic PD contexts [197]. Osmotin modulates autophagy via the AMPK/mTOR pathway and enhances dopaminergic neuron activity through Nurr1-dependent transcription. Its dual approach, targeting both inflammation and α-synuclein clearance, underscores its therapeutic potential [253]. Moreover, β-asarone, a distinct chemical compound, was identified to alleviate depression in PD by activating the PI3K/Akt/mTOR pathway, inhibiting autophagy, and reinstating synaptic activity in the hippocampus. Its neuroprotective and antidepressant properties indicate its potential application for concomitant PD symptoms [41]. Phloretin (PLT), a phenolic molecule, modulates the mTOR/NRF2/p62 pathway to enhance antioxidant defenses and reduce apoptosis and α-synuclein accumulation in rotenone-induced models, thereby substantiating the antioxidant-autophagy interface as a target for PD treatment [254]. Geniposide, derived from Fructus Gardeniae, improves motor performance and dopaminergic survival by mitigating oxidative stress through Nrf2 activation and mTOR-mediated apoptotic signaling, illustrating its dual effectiveness in cellular stress responses [255]. Crocin, a carotenoid extracted from saffron, activates the PI3K/Akt/mTOR pathway, enhancing neurotrophic indicators and reducing oxidative and apoptotic mediators. Its power to regulate miRNA-7 and miRNA-221 further solidifies its role as an autophagy regulator and its capability to protect neurons [256]. Ursodeoxycholic acid (UDCA) reinstates mitochondrial activity and autophagic flux through the AMPK/mTOR and PINK1/Parkin pathways. It concurrently enhances oxidative stress and mitigates dopaminergic neuronal loss in MPTP/MPP+ PD mice [257]. Asiatic acid (AA), derived from Centella asiatica, mitigates MPTP/probenecid-induced neurotoxicity by activating the PI3K/Akt/mTOR signaling pathway and inhibiting the MAPK/P38 axis, demonstrating a robust dual strategy for the survival of dopaminergic neurons [258]. Additionally, taurochenodeoxycholic acid (TCDCA), a component of bile acid, regulates neuroinflammation and mitochondrial dysfunction via AMPK/mTOR, Pink1/Parkin, and AKT/NFκB signaling pathways. Activation of the TGR5 receptor is essential for its diverse neuroprotective effects [260]. Lycium barbarum polysaccharide (LBP) also offers neuroprotection by augmenting antioxidant enzyme production. It reduces α-synuclein aggregation and activates the PTEN/AKT/mTOR pathway, hence preserving nigrostriatal integrity and cognitive function [261]. However, Cannabidiol (CBD) demonstrates in vitro-neuroprotection in vitro neuroprotection via ERK and AKT/mTOR pathways, mitigating apoptosis and autophagy dysregulation. Its interaction with CB2 and TRPV1 receptors further highlights it as a multi-receptor modulator in PD [262]. Furthermore, Idebenone, a coenzyme Q10 analogue with recognized antioxidant characteristics, was investigated for its possible involvement in PD through network pharmacology and cellular models. The study identified 87 possible targets, primarily associated with the MAP kinase and PI3K-AKT pathways. Molecular docking revealed substantial interactions with AKT and MAPK, whereas in vitro investigations confirmed that Idebenone induces autophagy and destroys α-synuclein via AKT/mTOR inhibition, hence affirming its therapeutic potential [263]. In addition to natural compounds, regulatory RNAs and transcription factors have demonstrated significant regulatory roles in modulating the mTOR pathway in PD. Programmed cell death protein 4 (PDCD4) is an elevated factor in PD and participates in neuronal apoptosis. Suppressing PDCD4 enhances the PI3K/AKT/mTOR pathway, leading to improved mitochondrial integrity and cellular viability [283]. Furthermore, Decoction of Rehmanniae (DOR) enhances PI3K/Akt/mTOR signaling to counteract MPP⁺-induced cytotoxicity, underscoring its role in apoptosis inhibition and oxidative stress mitigation [264]. L-DOPA, the cornerstone of PD treatment, also modulates mTOR signaling. It restores disrupted circadian rhythms and increases BMAL1 and CLOCK gene through the D1R-ERK1/2-mTOR pathway [265]. Additionally, long-term L-DOPA administration causes dyskinesia, which 5-hydroxytryptophan (5-HTP) suppresses by modulation of the hyperactivated striatal AKT/mTOR/S6K and CREB/ΔFosB pathway without compromising motor benefits [266]. Despite the scarcity of effective drugs for PD, recent studies have explored novel therapeutic compounds targeting important cellular processes. A study employed a chronic PD monkey model to evaluate the impact of Clioquinol (CQ). CQ significantly improved both motor and non-motor functions by reducing iron deposition and ROS in the SN, indicating the involvement of ferroptosis in its action. CQ also activated the AKT/mTOR pathway and inhibited p53-induced apoptosis, highlighting its neuroprotective potential [267]. Additionally, Hydrogen-saturated saline (HS) alleviated cardiovascular and motor symptoms during early and intermediate stages of rotenone-induced PD in rats by protecting RVLM catecholaminergic and nigral DA neurons. HS lowered ROS and α-synuclein, increased autophagy (through LC3 conversion, p62 degradation, and upregulation of ATG5 and Beclin-1), and inhibited the PI3K-Akt-mTOR pathway. Further, the absence of all these effects in late-stage PD rats suggests that HS primarily exerts neuroprotection before the development of significant motor impairments [284]. Further long non-coding RNA (lncRNA) and multiple microRNAs (miRNAs) have been implicated in PD pathogenesis through their influence on mTOR signaling and autophagy regulation. LncRNA SNHG1, for example, is overexpressed in PD and promotes autophagy inhibition through the miR-221/222/p27/mTOR pathway. Its downregulation restores autophagic markers and cell viability [285]. Among microRNAs, miR-199a also controls autophagy and neuronal survival by affecting the PTEN/AKT/mTOR pathway. Downregulation of miR-199a promotes autophagy and cell death while overexpression reverses these effects [286]. miR-410 and miR-181b protect DA neurons through regulation of the PTEN/AKT/mTOR signaling pathway, thereby maintaining autophagic equilibrium and cellular homeostasis [287, 288]. Similarly, miR-185 and miR-124 promote DA neuron survival through regulation of AMPK/mTOR-mediated autophagy and apoptosis [289, 290]. A further study contributed to the expanding evidence by examining the regulatory role of miR-410 in a 6-OHDA-induced cellular model of PD. Overexpression of miR-410 mitigated neuronal damage caused by 6-OHDA by decreasing apoptosis, ROS generation, and caspase-3 activity. These protective effects were facilitated by the direct targeting of PTEN, thus activating the AKT/mTOR pathway. The reversal of these benefits by PTEN overexpression validates the pivotal role of the PTEN/AKT/mTOR pathway in the neuroprotective function of miR-410 [287]. Complementary therapies also influence mTOR signaling with moxibustion and acupuncture, which have demonstrated efficacy in rotenone and DPD models, respectively. Moxibustion has been shown to enhance autophagic clearance of α-synuclein and boost behavioral outcomes in a rat model of PD due to suppression of the mTOR/p70S6K signaling pathway [291]. Recent research indicated that acupuncture mitigated motor and depressive symptoms in DPD model rats, elevated striatal levels of dopamine and serotonin, and reduced α-synuclein buildup. Furthermore, it also inhibited the autophagy in the striatum, enhanced the expression of phosphorylated mTOR, and thereby promoted synaptic repair. These results suggest that acupuncture's therapeutic effects in the DPD model mice may stem from modulating the mTOR signaling pathway, inhibiting autophagy-related α-syn clearance, and promoting synaptic repair [292]. These diverse findings collectively emphasize the therapeutic potential of regulating the AKT/mTOR pathway, via small chemicals, natural substances, or genetic manipulation, as an innovative approach for treating PD.
mTOR modulation in Huntington’s disease
Like AD and PD, the modulation of the mTOR signaling pathway using natural products, pharmacological agents, or inhibitors has shown therapeutic promise in the treatment of HD. HD is defined by the deleterious buildup of mHTT, which generates intracellular inclusion bodies and impairs neuronal function. A possible therapeutic approach in HD entails augmenting autophagy to promote the elimination of mHTT aggregates. In the initial phases of HD, autophagy is effectively preserved; however, as the disease advances, autolysosomal clearance becomes compromised. Research utilizing the Q175 HD mouse model in conjunction with an autophagy reporter line demonstrated the buildup of mHTT within autophagic vacuoles and the dysregulation of autophagy-related markers. Treatment with a mTOR inhibitor in the early stages of disease notably restored lysosomal function and diminished neuropathological characteristics, underscoring the therapeutic potential of autophagy activation as an early intervention method [42]. Moreover, recent research has illustrated the neuroprotective benefits of Morin hydrate (MH), a flavonoid with antioxidant characteristics, in a rat model of HD induced by 3-nitropropionic acid (3-NP). MH therapy markedly reduced motor impairments, weight reduction, and striatal neurodegeneration. Mechanistically, MH diminished the mTOR/IRE1-α, JNK/IP3R, and cytochrome c/caspase-3 pathways, while augmenting neuroprotective markers. The co-administration of WAG-4S, an ER stress inducer, partially mitigated the effects of MH, corroborating its mechanism through ER stress and mitochondrial regulation. Molecular docking also validated MH's affinity for mTOR, JNK, and IP3R, indicating a multi-target mechanism of action [268]. Additional evidence supporting the modulation of the mTOR pathway in HD is obtained from studies involving erythropoietin (EPO) and the Bacillus Calmette–Guérin (BCG) immunization. In a 3-NP-induced HD rat model, the administration of EPO, BCG, or their combination improved behavioral and histological outcomes. The neurotoxic effects of 3-NP, including oxidative stress, mitochondrial dysfunction, and the activation of the PI3K/Akt/mTOR/P70S6K signaling pathway, led to the inhibition of autophagy. EPO and BCG therapy rectified these changes, with the combination treatment demonstrating the most significant neuroprotective effects, highlighting their synergistic potential [269]. Further, CTEP, a selective mGluR5 inhibitor, has shown efficacy in mitigating HD pathogenesis in zQ175 mice. CTEP enhanced ULK1 activation via regulating dysregulated PI3K/Akt/mTOR signaling, hence promoting autophagy and reducing mHTT aggregates. Moreover, it augmented CREB-mediated synthesis of BDNF, hence facilitating neuronal survival and reducing apoptosis. These findings underscore the effectiveness of targeting mTOR-related pathways for the treatment of HD [270]. Given the adverse effects of extended mTOR inhibition with agents such as rapamycin, alternative methods to stimulate autophagy are under investigation. FDA-sanctioned pharmaceuticals, including L-type Ca²⁺ channel blockers, minoxidil, and clonidine, have shown efficacy in enhancing autophagy via an mTOR-independent pathway involving cAMP, IP3, calpain, and Gsα signaling. Elevated intracellular Ca²⁺, noted for its suppressive impact on autophagy and protein degradation, was also investigated in these animals, revealing other targets for pharmacological intervention [293]. Moreover, the combination of lithium, an IMPase inhibitor, with rapamycin has shown improved neuroprotection in HD fly models. Lithium promotes mTOR-independent autophagy but simultaneously stimulates mTOR via GSK-3β suppression, thereby undermining its advantages. The combined therapy significantly activates both mTOR-dependent and -independent autophagy pathways, exceeding the efficacy of singular therapies [294]. Nanotechnology has also entered the therapeutic realm. Nano-formulated ivabradine (nano-IVA) has recently demonstrated considerable efficacy in a 3-NP-induced HD rat model. Nano-IVA enhanced motor, cognitive, and psychiatric functions while regulating Rhes/mTOR-mediated autophagy. It diminished the expression of Rhes, mTOR, and Bcl-2, reinstated neurotransmitter levels, and maintained striatal histoarchitecture. These findings corroborate the efficacy of nano-IVA in addressing HD pathophysiology via molecular and cellular processes [271].
mTOR modulation in Amyotrophic Lateral Sclerosis
Similar to HD, regulation of the mTOR signaling pathway is widely acknowledged as a vital approach in the treatment of ALS, a progressive neurological disorder with few therapeutic alternatives. Niclosamide, an anti-fibrotic and anti-inflammatory compound, effectively delays disease progression, improving neurological outcomes, and extends the survival in SOD1-G93A and FUS-ALS mouse models. Its neuroprotective characteristics are associated with reduced neuroinflammation, gliosis, alongside the regulation of the STAT3 and mTOR pathways, decreased muscle atrophy, establishing it as a multi-target therapeutic alternative [272]. Additionally, oleanolic acid (OLA), a triterpenoid sourced from Olea europaea, was demonstrated that in mice with methylmercury-induced ALS for therapeutic benefits. It restored myelin and neurofilament protein levels, diminished inflammation, and improved performance, particularly when combined with edaravone. The advantages were achieved by suppressing the overactive PI3K/Akt/mTOR/STAT3/GSK-3β signaling pathway, underscoring its potential as a neuroprotective agent [273]. Similarly, ibrutinib, a Bruton's tyrosine kinase (BTK) inhibitor, was evaluated for potential therapeutic efficacy in SOD1-G93A models by reducing both muscle atrophy and inflammation. Oral ibrutinib postponed symptom emergence, prolonged lifetime, and enhanced motor function. It diminished pro-inflammatory cytokines and glial markers (IBA-1 and GFAP) while reinstating the muscle-to-body weight ratio. These effects were linked to the regulation of the PI3K/Akt/mTOR signaling pathway in both the brain and spinal cord [274]. Environmental variables such as methylmercury (MeHg⁺) exposure are associated with the etiology of ALS, primarily through the destruction of oligodendrocytes and the loss of myelin. In rats exposed to MeHg⁺, guggulsterone (GST), a substance obtained from Commiphora resin, reinstated motor and cognitive functions, improved remyelination, and diminished neuroinflammation. GST influenced dysregulated STAT3, mTOR, and PPAR-γ pathways and showed synergistic advantages when administered with vitamin D3 [275]. Furthermore, astrocyte reactivity, induced by IGF1R-mediated mTOR activation, is crucial in familial ALS associated with mutant SOD1. Aberrant mTOR activation inhibits autophagy and promotes astrocyte growth and toxicity, leading to motor neuron death. Inhibition of the IGF1R-mTOR axis has demonstrated a reduction in astrocyte-mediated neurotoxicity, presenting a viable therapeutic target [240]. At the post-transcriptional level, microRNA-193b-3p has become a critical regulator of mTOR signaling in ALS. It is significantly downregulated in both ALS patients and in SOD1G93A mice. By specifically targeting TSC1, the downregulation of miR-193b-3p results in the accumulation of TSC1, inactivation of mTORC1, increased autophagy, and higher cell survival. Conversely, its overexpression suppresses autophagy and promotes cell death. Consequently, the regulation of miR-193b-3p could offer a viable approach for mTOR-targeted therapy in ALS [296].
Conclusion
NDDs, such as AD, PD, HD, and ALS, are increasingly common due to rising global life expectancy, currently affecting about 30 million people. Despite their widespread impact and significant socio-economic burden, effective remedies are nonetheless constrained. The mTOR and its associated signaling pathways have garnered interest as a crucial regulator in NDs because of their involvement in autophagy, cellular proliferation, protein synthesis, and metabolism. In AD, mTOR dysregulation is associated with the accumulation of Aβ and hyperphosphorylated tau. Hyperactive mTOR suppresses autophagy, obstructing the removal of misfolded proteins and facilitating Aβ buildup. It also augments β- and γ-secretase activity, promoting the amyloidogenic processing of APP. mTOR facilitates tau disease by expediting its hyperphosphorylation and hindering breakdown. Pharmacological inhibition of mTOR, exemplified by rapamycin, has diminished Aβ and tau pathogenesis in experimental animals. Nonetheless, mTOR demonstrates a dual function. Chronic overactivation leads to neurodegeneration through disrupted proteostasis and neuroinflammation, but short-term or context-specific activation can enhance neuronal survival and repair, in part by diminishing apoptosis produced by immune cells. This contradiction underscores the intricacy of targeting mTOR in NDs. Therapeutically, partial mTOR inhibition may diminish harmful protein buildup, restore autophagic function, and maintain neuronal integrity. However, excessive inhibition may hinder neuronal development and cognitive function. The interplay of mTOR with pathways such as GSK-3β, PI3K/Akt, AMPK, and insulin/IGF-1 complicates the development of selective, low-toxicity inhibitors. Preclinical investigations with rapamycin and analogous substances demonstrate potential in enhancing cognitive function and decelerating illness progression. Future strategies should emphasize precise, temporal, and tissue-specific regulation of mTOR, potentially in conjunction with other pathway targets, to augment treatment efficacy while reducing side effects. Future investigations also require moving toward delineating the complex and dynamic cross-talk between mTOR signaling and other critical cellular systems to broaden the translational dimension of this field. A primary focus should be the intricate relationship between mTOR and mitochondrial dynamics, specifically, how sustained mTORC1 hyperactivity compromises the delicate balance of mitochondrial fission, fusion, and mitophagy, accelerating neuronal bioenergetic collapse. Concurrently, deeper study into neuroinflammatory signaling is warranted, as understanding how mTOR regulates microglial and astrocytic immunometabolism, including their shift between neuroprotective and reactive states, will be essential for target specificity. Moreover, dissecting mTOR's role in glial metabolism, particularly in astrocytic lactate shuttling and nutrient trafficking. This research could lead to interventions that restore the metabolic symbiosis critical for neuronal survival and function. By integrating these systems-level views, researchers can transition from solely targeting mTOR to developing multi-faceted, metabolic interventions that simultaneously address proteotoxicity, energy deficits, and neuroinflammation.
Acknowledgements
Author contributions NKJ conceptualized the manuscript. NKJ, PC, MMA, DA, AGA, SL, AS, GMS, HAA, HAM, JM, RG, KT, and DK performed the literature survey and drafted and edited the manuscript. NKJ ideated the scheme and performed the artwork. All authors contributed to the article and approved the submitted version.
Consent for publication All the authors read and approved the final manuscript.
Disclosure Statement
The authors declare no competing interests.
References
- Perluigi M, Di Domenico F, Barone E, Butterfield DA. mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder. Free Radic Biol Med. 2021;169:382-96. https://doi.org/10.1016/j.freeradbiomed.2021.04.025
- Perluigi M, Di Domenico F, Butterfield DA. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis. 2015;84:39-49. https://doi.org/10.1016/j.nbd.2015.03.014
- Choo AY, Yoon SO, Sang GK, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105(45):17414-9. https://doi.org/10.1073/pnas.0809136105
- Ma Y, Vassetzky Y, Dokudovskaya S. mTORC1 pathway in DNA damage response. Biochim Biophys Acta Mol Cell Res. 2018;1865(9):1293-311. https://doi.org/10.1016/j.bbamcr.2018.06.011
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122-8. https://doi.org/10.1038/ncb1183
- Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20(20):2820-32. https://doi.org/10.1101/gad.1461206
- Steinmetz JD, Seeher KM, Schiess N, Nichols E, Cao B, Servili C, et al. Global, regional, and national burden of disorders affecting the nervous system, 1990-2021: a systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 2024;23(4):344-81. https://doi.org/10.1016/S1474-4422(24)00038-3
- Breijyeh Z, Karaman R. Comprehensive Review on Alzheimer's Disease: Causes and Treatment. Molecules. 2020;25:5789.https://doi.org/10.3390/molecules25245789
- Melick CH, Jewell JL. Regulation of mTORC1 by Upstream Stimuli. Genes 2020, Vol 11, Page 989. 2020;11(9):989. https://doi.org/10.3390/genes11090989
- Hodges SL, Reynolds CD, Smith GD, Jefferson TS, Nolan SO, Lugo JN. Molecular interplay between hyperactive mammalian target of rapamycin signaling and Alzheimer's disease neuropathology in the NS-Pten knockout mouse model. Neuroreport. 2018;29(13):1109-13. https://doi.org/10.1097/WNR.0000000000001081
- Martín-Flores N, Pérez-Sisqués L, Creus-Muncunill J, Masana M, Ginés S, Alberch J, et al. Synaptic RTP801 contributes to motor-learning dysfunction in Huntington's disease. Cell Death & Disease. 2020;11(7):1-15. https://doi.org/10.1038/s41419-020-02775-5
- Nakka VP, Prakash-babu P, Vemuganti R. Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries. Mol Neurobiol. 2016;53(1):532-44. https://doi.org/10.1007/s12035-014-9029-6
- Maiese K. Taking aim at Alzheimer's disease through the mammalian target of rapamycin. Ann Med. 2014;46(8):587. https://doi.org/10.3109/07853890.2014.941921
- Xie PL, Zheng MY, Han R, Chen WX, Mao JH. Pharmacological mTOR inhibitors in ameliorating Alzheimer's disease: current review and perspectives. Front Pharmacol. 2024;15:1366061. https://doi.org/10.3389/fphar.2024.1366061
- Goul C, Peruzzo R, Zoncu R. The molecular basis of nutrient sensing and signalling by mTORC1 in metabolism regulation and disease. Nature Reviews Molecular Cell Biology. 2023;24(12):857-75. https://doi.org/10.1038/s41580-023-00641-8
- Davoody S, Asgari Taei A, Khodabakhsh P, Dargahi L. mTOR signaling and Alzheimer's disease: What we know and where we are? CNS Neurosci Ther. 2024;30(4):e14463. https://doi.org/10.1111/cns.14463
- Narayanan L, Thekkekkara D, Kondaveeti SN, Babu A, Sailen BA, Chidambaram SB, et al. Deciphering the intricate role of mTOR signaling and autophagy in Parkinson's disease and therapeutic prospects. J Appl Pharm Sci. 2024;14,(3):001-10. https://doi.org/10.7324/JAPS.2023.156692
- Duong S, Patel T, Chang F. Dementia: What pharmacists need to know. Can Pharm J (Ott). 2017;150(2):118-29. https://doi.org/10.1177/1715163517690745
- Ding Y, Liu H, Cen M, Tao Y, Lai C, Tang Z. Rapamycin Ameliorates Cognitive Impairments and Alzheimer's Disease-Like Pathology with Restoring Mitochondrial Abnormality in the Hippocampus of Streptozotocin-Induced Diabetic Mice. Neurochem Res. 2021;46(2):265-75. https://doi.org/10.1007/s11064-020-03160-6
- Kaeberlein M, Galvan V. Rapamycin and Alzheimer's disease: Time for a clinical trial? Sci Transl Med. 2019;11(476):eaar4289. https://doi.org/10.1126/scitranslmed.aar4289
- Yu JJ, Goncharova EA. mTOR Signaling Network in Cell Biology and Human Disease. Int J Mol Sci. 2022;23(24):16142. https://doi.org/10.3390/ijms232416142
- Dazert E, Hall MN. MTOR signaling in disease. Curr Opin Cell Biol. 2011;23(6):744-55. https://doi.org/10.1016/j.ceb.2011.09.003
- Zhou H, Huang S. The complexes of mammalian target of rapamycin. Curr Protein Pept Sci. 2010;11(6):409. https://doi.org/10.2174/138920310791824093
- Cai H, Dong LQ, Liu F. Recent Advances in Adipose mTOR Signaling and Function: Therapeutic Prospects. Trends Pharmacol Sci. 2016;37(4):303-17. https://doi.org/10.1016/j.tips.2015.11.011
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell. 2002;110(2):177-89. https://doi.org/10.1016/S0092-8674(02)00833-4
- Hoeffer CA, Klann E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33(2):67-75. https://doi.org/10.1016/j.tins.2009.11.003
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mTORC1. Science. 2008;320(5882):1496-501.https://doi.org/10.1126/science.1157535
- Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: Signalling inputs, substrates and feedback mechanisms. Cell Signal. 2009;21(6):827-35. https://doi.org/10.1016/j.cellsig.2009.01.012
- Laplante M, Sabatini DM. MTOR signaling in growth control and disease. Cell. 2012;149(2):274-93.https://doi.org/10.1016/j.cell.2012.03.017
- Crino PB. MTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med. 2011;17(12):734-42. https://doi.org/10.1016/j.molmed.2011.07.008
- Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell. 2006;124(3):471-84. https://doi.org/10.1016/j.cell.2006.01.016
- Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 2014;7:28. https://doi.org/10.3389/fnmol.2014.00028
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21(4):183-203. https://doi.org/10.1038/s41580-019-0199-y
- Wei X, Luo L, Chen J. Roles of mTOR Signaling in Tissue Regeneration. Cells. 2019;8(9):1075. https://doi.org/10.3390/cells8091075
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55-61. https://doi.org/10.1038/nature10912
- Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10(1):31. https://doi.org/10.1186/s13578-020-00396-1
- Mao B., Zhang Q., Ma L., Zhao D-S., Zhao P., Yan P, et al. Overview of Research into mTOR Inhibitors. Molecules. 2022;27(16):5295. https://doi.org/10.3390/molecules27165295
- Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144(5):757-68. https://doi.org/10.1016/j.cell.2011.02.014
- Tian T, Li X, Zhang J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int J Mol Sci. 2019;20(3):755. https://doi.org/10.3390/ijms20030755
- Sanganahalli BG, Mihailovic JM, Vekaria HJ, Coman D, Yackzan AT, Flemister A, et al. mTOR inhibition enhances synaptic and mitochondrial function in Alzheimer's disease in an APOE genotype-dependent manner. Journal of Cerebral Blood Flow & Metabolism. 2024;44(10):1745-58. https://doi.org/10.1177/0271678X241261942
- Wang Z, Huang P, Wang N, Zhang Q, Kang J, Fang Y, et al. β-asarone inhibits autophagy by activating the PI3K/Akt/mTOR pathway in a rat model of depression in Parkinson's disease. Behavioural Brain Research. 2024;465:114966. https://doi.org/10.1016/j.bbr.2024.114966
- Stavrides P, Goulbourne CN, Peddy J, Huo C, Rao M, Khetarpal V, et al. mTOR inhibition in Q175 Huntington's disease model mice facilitates neuronal autophagy and mutant huntingtin clearance. Elife. 2025;14:RP104979. https://doi.org/10.7554/eLife.104979
- Liu X, Guo B, Li Q, Nie J. mTOR in metabolic homeostasis and disease. Exp Cell Res. 2024;441(2):114173. https://doi.org/10.1016/j.yexcr.2024.114173
- Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nature Reviews Immunology. 2012;12(5):325-38. https://doi.org/10.1038/nri3198
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature. 2017;552(7685):368-73. https://doi.org/10.1038/nature25023
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5(6):566-71. https://doi.org/10.1038/ncb996
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648-57. https://doi.org/10.1038/ncb839
- Wataya-Kaneda M. Mammalian target of rapamycin and tuberous sclerosis complex. J Dermatol Sci. 2015;79(2):93-100. https://doi.org/10.1016/j.jdermsci.2015.04.005
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658-65. https://doi.org/10.1038/ncb840
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proceedings of the National Academy of Sciences. 2004;101(37):13489-94.https://doi.org/10.1073/pnas.0405659101
- Carrière A, Cargnello M, Julien LA, Gao H, Bonneil É, Thibault P, et al. Oncogenic MAPK Signaling Stimulates mTORC1 Activity by Promoting RSK-Mediated Raptor Phosphorylation. Current Biology. 2008;18(17):1269-77.https://doi.org/10.1016/j.cub.2008.07.078
- Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, et al. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J Biol Chem. 2011;286(1):567-77.https://doi.org/10.1074/jbc.M110.159046
- Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J Immunol. 2017;198(3):1006-14.https://doi.org/10.4049/jimmunol.1601515
- Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27(4):286-96.https://doi.org/10.1016/j.smim.2015.08.001
- Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, et al. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proceedings of the National Academy of Sciences. 2012;109(1):273-8.https://doi.org/10.1073/pnas.1118579109
- Troutman TD, Bazan JF, Pasare C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle. 2012;11(19):3559-67.https://doi.org/10.4161/cc.21572
- Inoki K, Zhu T, Guan KL. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell. 2003 Nov;115(5):577-90.https://doi.org/10.1016/S0092-8674(03)00929-2
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol Cell. 2008;30(2):214-26.https://doi.org/10.1016/j.molcel.2008.03.003
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell. 2010;141(2):290-303.https://doi.org/10.1016/j.cell.2010.02.024
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340(6136):1100-6.https://doi.org/10.1126/science.1232044
- Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The sestrins interact with gator2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9(1):1-8.https://doi.org/10.1016/j.celrep.2014.09.014
- Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, et al. Sestrins Inhibit mTORC1 Kinase Activation through the GATOR Complex. Cell Rep. 2014;9(4):1281-91.https://doi.org/10.1016/j.celrep.2014.10.019
- Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43-8.https://doi.org/10.1126/science.aab2674
- Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell. 2016;165(1):153-64.https://doi.org/10.1016/j.cell.2016.02.035
- Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai L V., et al. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell. 2017;171(3):642-654.e12.https://doi.org/10.1016/j.cell.2017.09.046
- Wang S, Tsun Z-Y, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347(6218):188-94.https://doi.org/10.1126/science.1257132
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, et al. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347(6218):194-8.https://doi.org/10.1126/science.1259472
- Durán R V., Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis Activates Rag-mTORC1 Signaling. Mol Cell. 2012;47(3):349-58.https://doi.org/10.1016/j.molcel.2012.05.043
- Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns(3, 4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015;5(11):1194-11209.https://doi.org/10.1158/2159-8290.CD-15-0460
- Kazyken D, Magnuson B, Bodur C, Acosta-Jaquez HA, Zhang D, Tong X, et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci Signal. 2019;12(585):eaav3249.https://doi.org/10.1126/scisignal.aav3249
- Domenico F Di, Head E, Butterfield DA, Perluigi M. Oxidative Stress and Proteostasis Network: Culprit and Casualty of Alzheimer's-Like Neurodegeneration. Adv Geriatr. 2014;2014(1):527518.https://doi.org/10.1155/2014/527518
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24-49.https://doi.org/10.1016/j.pneurobio.2013.10.004
- Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med. 2013;62:170-85.https://doi.org/10.1016/j.freeradbiomed.2012.09.016
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069-75.https://doi.org/10.1038/nature06639
- Nelson VK, Begum MY, Suryadevara PR, Madhuri Kallam SD, Panda SP, Bodapati A, et al. Natural bioactive compounds as modulators of autophagy: A herbal approach to the management of neurodegenerative diseases. Eur J Pharmacol. 2025;1005:178003.https://doi.org/10.1016/j.ejphar.2025.178003
- Wojcik S. Crosstalk between autophagy and proteasome protein degradation systems: possible implications for cancer therapy. Folia Histochem Cytobiol. 2014;51(4):249-64.https://doi.org/10.5603/FHC.2013.0036
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124-31.https://doi.org/10.1016/j.ceb.2009.11.014
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol Rev. 2010;90(4):1383-435.https://doi.org/10.1152/physrev.00030.2009
- García-Arencibia M, Hochfeld WE, Toh PPC, Rubinsztein DC. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol. 2010;21(7):691-8.https://doi.org/10.1016/j.semcdb.2010.02.008
- Metcalf DJ, García-Arencibia M, Hochfeld WE, Rubinsztein DC. Autophagy and misfolded proteins in neurodegeneration. Exp Neurol. 2012;238(1):22.https://doi.org/10.1016/j.expneurol.2010.11.003
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095-108.https://doi.org/10.1038/emboj.2012.32
- Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7(11):1379-81.https://doi.org/10.4161/auto.7.11.17166
- Kist M, Vucic D. Cell death pathways: intricate connections and disease implications. EMBO J. 2021 Mar;40(5):e106700.https://doi.org/10.15252/embj.2020106700
- Han J, Wang L, Lv H, Liu J, Dong Y, Shi L, et al. EphA2 inhibits SRA01/04 cells apoptosis by suppressing autophagy via activating PI3K/Akt/mTOR pathway. Arch Biochem Biophys. 2021;711:109024.https://doi.org/10.1016/j.abb.2021.109024
- Liu W, Wang X, Liu Z, Wang Y, Yin B, Yu P, et al. SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway. Br J Cancer. 2017;117(8):1139-53.https://doi.org/10.1038/bjc.2017.293
- Gong X, Jiang L, Li W, Liang Q, Li Z. Curcumin induces apoptosis and autophagy inhuman renal cell carcinoma cells via Akt/mTOR suppression. Bioengineered. 2021;12(1):5017-27.https://doi.org/10.1080/21655979.2021.1960765
- Wang H, Bai G, Chen J, Han W, Guo R, Cui N. mTOR deletion ameliorates CD4 + T cell apoptosis during sepsis by improving autophagosome-lysosome fusion. Apoptosis. 2022;27(5-6):401-8.https://doi.org/10.1007/s10495-022-01719-y
- Wang R, Hu W. Asprosin promotes β‐cell apoptosis by inhibiting the autophagy of β‐cell via AMPK‐mTOR pathway. J Cell Physiol. 2021;236(1):215-21.https://doi.org/10.1002/jcp.29835
- Xu J, Deng Y, Wang Y, Sun X, Chen S, Fu G. SPAG5‐AS1 inhibited autophagy and aggravated apoptosis of podocytes via SPAG5/AKT/mTOR pathway. Cell Prolif. 2020;53(2):e12738.https://doi.org/10.1111/cpr.12738
- Hu X, Xia M, Wang J, Yu H, Chai J, Zhang Z, et al. Dual PI3K/mTOR inhibitor PKI-402 suppresses the growth of ovarian cancer cells by degradation of Mcl-1 through autophagy. Biomed Pharmacother. 2020;129:110397.https://doi.org/10.1016/j.biopha.2020.110397
- Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44(5):698-709.https://doi.org/10.1016/j.molcel.2011.10.014
- Han J, Hou W, Goldstein LA, Stolz DB, Watkins SC, Rabinowich H. A Complex between Atg7 and Caspase-9. Journal of Biological Chemistry. 2014;289(10):6485-97.https://doi.org/10.1074/jbc.M113.536854
- Girodengo M, Ultanir SK, Bateman JM. Mechanistic target of rapamycin signaling in human nervous system development and disease. Front Mol Neurosci. 2022;15:1005631.https://doi.org/10.3389/fnmol.2022.1005631
- Zhang J, Gan W, Peng R, Lu L, Lu W, Liu J. Activation of the mTOR pathway promotes neurite growth through upregulation of CD44 expression. Journal of International Medical Research. 2023;51(6):3000605231178510.https://doi.org/10.1177/03000605231178510
- Troca-Marin J, Casanas J, Benito I, Montesinos M. The Akt-mTOR Pathway in Down's Syndrome: The Potential Use of Rapamycin/Rapalogs for Treating Cognitive Deficits. CNS Neurol Disord Drug Targets. 2014;13(1):34-40.https://doi.org/10.2174/18715273113126660184
- Licausi F, Hartman NW. Role of mTOR Complexes in Neurogenesis. Int J Mol Sci. 2018;19(5):1544.https://doi.org/10.3390/ijms19051544
- O'Neill C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer's disease. Exp Gerontol. 2013;48(7):647-53.https://doi.org/10.1016/j.exger.2013.02.025
- Dwyer JM, Duman RS. Activation of mammalian target of rapamycin and synaptogenesis: Role in the actions of rapid-acting antidepressants. Biol Psychiatry. 2013;73(12):1189-98.https://doi.org/10.1016/j.biopsych.2012.11.011
- Russo E, Citraro R, Constanti A, De Sarro G. The mTOR Signaling Pathway in the Brain: Focus on Epilepsy and Epileptogenesis. Mol Neurobiol. 2012;46(3):662-81.https://doi.org/10.1007/s12035-012-8314-5
- Sosanya NM, Cacheaux LP, Workman ER, Niere F, Perrone-Bizzozero NI, Raab-Graham KF. Mammalian Target of Rapamycin (mTOR) Tagging Promotes Dendritic Branch Variability through the Capture of Ca2+/Calmodulin-dependent Protein Kinase II α (CaMKIIα) mRNAs by the RNA-binding Protein HuD. Journal of Biological Chemistry. 2015;290(26):16357-71.https://doi.org/10.1074/jbc.M114.599399
- Lau KA, Yang X, Rioult-Pedotti MS, Tang S, Appleman M, Zhang J, et al. A PSD-95 peptidomimetic mitigates neurological deficits in a mouse model of Angelman syndrome. Prog Neurobiol. 2023;230:102513.https://doi.org/10.1016/j.pneurobio.2023.102513
- Pena-Leon V, Perez-Lois R, Seoane LM. mTOR Pathway is Involved in Energy Homeostasis Regulation as a Part of the Gut-Brain Axis. Int J Mol Sci. 2020;21(16):5715.https://doi.org/10.3390/ijms21165715
- Crislip GR, Johnston JG, Douma LG, Costello HM, Juffre A, Boyd K, et al. Circadian Rhythm Effects on the Molecular Regulation of Physiological Systems. Compr Physiol. 2021;12(1):2769-98.https://doi.org/10.1002/j.2040-4603.2022.tb00198.x
- Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Translational Neurodegeneration. 2020;9(1):1-12.https://doi.org/10.1186/s40035-020-00221-2
- Almutary AG, Begum MY, Kyada AK, Gupta S, Jyothi SR, Chaudhary K, et al. Inflammatory signaling pathways in Alzheimer's disease: Mechanistic insights and possible therapeutic interventions. Ageing Res Rev. 2025;104:102548.https://doi.org/10.1016/j.arr.2024.102548
- Zeng C, Hu J, Chen F, Huang T, Zhang L. The Coordination of mTOR Signaling and Non-Coding RNA in Regulating Epileptic Neuroinflammation. Front Immunol. 2022;13:924642.https://doi.org/10.3389/fimmu.2022.924642
- Sugama S. Stress-induced microglial activation may facilitate the progression of neurodegenerative disorders. Med Hypotheses. 2009;73(6):1031-4.https://doi.org/10.1016/j.mehy.2009.02.047
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20(16):6309-16.https://doi.org/10.1523/JNEUROSCI.20-16-06309.2000
- Tringali G, Pozzoli G, Vairano M, Mores N, Preziosi P, Navarra P. Interleukin-18 displays effects opposite to those of interleukin-1 in the regulation of neuroendocrine stress axis. J Neuroimmunol. 2005;160(1-2):61-7.https://doi.org/10.1016/j.jneuroim.2004.10.028
- Galea E, Feinstein DL, Reis DJ. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proceedings of the National Academy of Sciences. 1992;89(22):10945-9.https://doi.org/10.1073/pnas.89.22.10945
- Zhang Z, Wang X, Zhang D, Liu Y, Li L. Geniposide-mediated protection against amyloid deposition and behavioral impairment correlates with downregulation of mTOR signaling and enhanced autophagy in a mouse model of Alzheimer's disease. Aging. 2019;11(2):536-48.https://doi.org/10.18632/aging.101759
- Schwartz RH. T Cell Anergy. Annu Rev Immunol. 2003;21(1):305-34.https://doi.org/10.1146/annurev.immunol.21.120601.141110
- Powell JD, Delgoffe GM. The Mammalian Target of Rapamycin: Linking T Cell Differentiation, Function, and Metabolism. Immunity. 2010;33(3):301-11.https://doi.org/10.1016/j.immuni.2010.09.002
- Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T Cells Are Metabolically Anergic. The Journal of Immunology. 2009;183(10):6095-101.https://doi.org/10.4049/jimmunol.0803510
- Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. Journal of Experimental Medicine. 2009;206(13):3015-29.https://doi.org/10.1084/jem.20090847
- Vashisth K, Sharma S, Ghosh S, Babu MA, Ghosh S, Iqbal D, et al. Immunotherapy in Alzheimer's Disease: Current Status and Future Directions. Journal of Alzheimer's Disease. 2024;101(s1):S23-39.https://doi.org/10.3233/JAD-230603
- Sethi P, Bhaskar R, Singh KK, Gupta S, Han SS, Avinash D, et al. Exploring advancements in early detection of Alzheimer's disease with molecular assays and animal models. Ageing Res Rev. 2024;100:102411.https://doi.org/10.1016/j.arr.2024.102411
- Kumar Nelson V, Jha NK, Nuli MV, Gupta S, Kanna S, Gahtani RM, et al. Unveiling the impact of aging on BBB and Alzheimer's disease: Factors and therapeutic implications. Ageing Res Rev. 2024;98:102224.https://doi.org/10.1016/j.arr.2024.102224
- Albadrani HM, Chauhan P, Ashique S, Babu MA, Iqbal D, Almutary AG, et al. Mechanistic insights into the potential role of dietary polyphenols and their nanoformulation in the management of Alzheimer's disease. Biomed Pharmacother. 2024;174:116376.https://doi.org/10.1016/j.biopha.2024.116376
- Plascencia-Villa G, Perry G. Neuropathologic Changes Provide Insights into Key Mechanisms of Alzheimer Disease and Related Dementia. American Journal of Pathology. 2022;192(10):1340-6.https://doi.org/10.1016/j.ajpath.2022.07.002
- Guo J, Xue J, Ding Z, Li X, Wang X, Xue H. Activated Phosphoinositide 3-Kinase/Akt/Mammalian Target of Rapamycin Signal and Suppressed Autophagy Participate in Protection Offered by Licochalcone A Against Amyloid-β Peptide Fragment 25-35-Induced Injury in SH-SY5Y Cells. World Neurosurg. 2022;157:e390-400.https://doi.org/10.1016/j.wneu.2021.10.098
- Shahani N, Pryor W, Swarnkar S, Kholodilov N, Thinakaran G, Burke RE, et al. Rheb GTPase Regulates β-Secretase Levels and Amyloid β Generation. Journal of Biological Chemistry. 2014;289(9):5799-808.https://doi.org/10.1074/jbc.M113.532713
- Vassar R, Kandalepas PC. The β-secretase enzyme BACE1 as a therapeutic target for Alzheimer's disease. Alzheimers Res Ther. 2011;3(3):20.https://doi.org/10.1186/alzrt82
- Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 2005;272(16):4211-20.https://doi.org/10.1111/j.1742-4658.2005.04833.x
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Current Biology. 2006;16(3):230-41.https://doi.org/10.1016/j.cub.2005.12.042
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: Neurodegeneration Without Neurofibrillary Tangles. Science. 2001;293(5530):711-4.https://doi.org/10.1126/science.1062382
- Tang Z, Ioja E, Bereczki E, Hultenby K, Li C, Guan Z, et al. mTor mediates tau localization and secretion: Implication for Alzheimer's disease. Biochim Biophys Acta Mol Cell Res. 2015;1853(7):1646-57.https://doi.org/10.1016/j.bbamcr.2015.03.003
- Tang Z, Bereczki E, Zhang H, Wang S, Li C, Ji X, et al. Mammalian Target of Rapamycin (mTor) Mediates Tau Protein Dyshomeostasis. Journal of Biological Chemistry. 2013;288(22):15556-70.https://doi.org/10.1074/jbc.M112.435123
- Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A. 2010;107(50):21830-5.https://doi.org/10.1073/pnas.0912793107
- Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer's disease. J Cell Mol Med. 2008;12(6b):2525.https://doi.org/10.1111/j.1582-4934.2008.00509.x
- Sun Y-X, Ji X, Mao X, Xie L, Jia J, Galvan V, et al. Differential Activation of mTOR Complex 1 Signaling in Human Brain with Mild to Severe Alzheimer's Disease. Journal of Alzheimer's Disease. 2013;38(2):437-44.https://doi.org/10.3233/JAD-131124
- Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, et al. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. 2005;93(1):105-17.https://doi.org/10.1111/j.1471-4159.2004.02949.x
- Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, et al. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre‐clinical AD, amnestic mild cognitive impairment and late‐stage AD. J Neurochem. 2015;133(5):739-49.https://doi.org/10.1111/jnc.13037
- Zheng M, Wang P. Role of insulin receptor substance-1 modulating PI3K/Akt insulin signaling pathway in Alzheimer's disease. 3 Biotech. 2021;11(4):179.https://doi.org/10.1007/s13205-021-02738-3
- Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem. 2005;94(1):215-25.https://doi.org/10.1111/j.1471-4159.2005.03187.x
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285(17):13107-20.https://doi.org/10.1074/jbc.M110.100420
- Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magrí A, et al. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem. 2011;286(11):8924-32.https://doi.org/10.1074/jbc.M110.180638
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol Cell. 2007;25(6):903-15.https://doi.org/10.1016/j.molcel.2007.03.003
- François A, Terro F, Janet T, Bilan AR, Paccalin M, Page G. Involvement of interleukin-1β in the autophagic process of microglia: relevance to Alzheimer's disease. J Neuroinflammation. 2013;10(1):915.https://doi.org/10.1186/1742-2094-10-151
- Damjanac M, Page G, Ragot S, Laborie G, Gil R, Hugon J, et al. PKR, a cognitive decline biomarker, can regulate translation via two consecutive molecular targets p53 and Redd1 in lymphocytes of AD patients. J Cell Mol Med. 2009;13(8B):1823-32.https://doi.org/10.1111/j.1582-4934.2009.00688.x
- Morel M, Couturier J, Pontcharraud R, Gil R, Fauconneau B, Paccalin M, et al. Evidence of molecular links between PKR and mTOR signalling pathways in Aβ neurotoxicity: Role of p53, Redd1 and TSC2. Neurobiol Dis. 2009;36(1):151-61.https://doi.org/10.1016/j.nbd.2009.07.004
- Zhang F, Beharry ZM, Harris TE, Lilly MB, Smith CD, Mahajan S, et al. PIM1 Protein Kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol Ther. 2009;8(9):846-53.https://doi.org/10.4161/cbt.8.9.8210
- Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4(1):14.https://doi.org/10.1186/1750-1326-4-14
- Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci (Schol Ed). 2012;4(3):941.https://doi.org/10.2741/s310
- Damjanac M, Rioux Bilan A, Paccalin M, Pontcharraud R, Fauconneau B, Hugon J, et al. Dissociation of Akt/PKB and ribosomal S6 kinase signaling markers in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2008;29(2):354-67.https://doi.org/10.1016/j.nbd.2007.09.008
- Njie e. MG, Boelen E, Stassen FR, Steinbusch HWM, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging. 2012;33(1):195.e1-195.e12.https://doi.org/10.1016/j.neurobiolaging.2010.05.008
- Browne TC, McQuillan K, McManus RM, O'Reilly J-A, Mills KHG, Lynch MA. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease. J Immunol. 2013;190(5):2241-51.https://doi.org/10.4049/jimmunol.1200947
- McManus RM, Higgins SC, Mills KHG, Lynch MA. Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol Aging. 2014;35(1):109-21.https://doi.org/10.1016/j.neurobiolaging.2013.07.025
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity. 2009;30(6):832-44.https://doi.org/10.1016/j.immuni.2009.04.014
- Katholnig K, Linke M, Pham H, Hengstschläger M, Weichhart T. Immune responses of macrophages and dendritic cells regulated by mTOR signalling. Biochem Soc Trans. 2013;41(4):927-33.https://doi.org/10.1042/BST20130032
- Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of Immune Responses by mTOR. Annu Rev Immunol. 2012;30(1):39-68.https://doi.org/10.1146/annurev-immunol-020711-075024
- Yates SC, Zafar A, Hubbard P, Nagy S, Durant S, Bicknell R, et al. Dysfunction of the mTOR pathway is a risk factor for Alzheimer's disease. Acta Neuropathol Commun. 2013;1(1):3.https://doi.org/10.1186/2051-5960-1-3
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983-97.https://doi.org/10.1038/nm.3232
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120(Pt 23):4081-91.https://doi.org/10.1242/jcs.019265
- Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, et al. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: Implications for β-amyloid peptide over-production and localization in Alzheimer's disease. International Journal of Biochemistry and Cell Biology. 2004;36(12):2531-40.https://doi.org/10.1016/j.biocel.2004.05.010
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee J-H, et al. Macroautophagy-a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171(1):87-98.https://doi.org/10.1083/jcb.200505082
- Nilsson P, Saido TC. Dual roles for autophagy: degradation and secretion of Alzheimer's disease Aβ peptide. Bioessays. 2014;36(6):570-8.https://doi.org/10.1002/bies.201400002
- Woods NK, Padmanabhan J. Neuronal Calcium Signaling and Alzheimer's Disease. Advances in Experimental edicine and biology. 2012;740:1193-217.https://doi.org/10.1007/978-94-007-2888-2_54
- Ma T. GSK3 in Alzheimer's disease: mind the isoforms. J Alzheimers Dis. 2014;39(4):707-10.https://doi.org/10.3233/JAD-131661
- Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP‐activated protein kinase: a potential player in Alzheimer's disease. J Neurochem. 2011;118(4):460-74.https://doi.org/10.1111/j.1471-4159.2011.07331.x
- Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010;5(6):e11102.https://doi.org/10.1371/journal.pone.0011102
- McBrayer M, Nixon RA. Lysosome and calcium dysregulation in Alzheimer's disease: partners in crime. Biochem Soc Trans. 2013;41(6):1495-502.https://doi.org/10.1042/BST20130201
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. Journal of Clinical Investigation. 2008;118(6):2190-9.https://doi.org/10.1172/JCI33585
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-β Levels in a Mouse Model of Alzheimer's Disease. PLoS One. 2010;5(4):e9979.https://doi.org/10.1371/journal.pone.0009979
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288(2):1295-306.https://doi.org/10.1074/jbc.M112.409250
- Liu D, Pitta M, Jiang H, Lee JH, Zhang G, Chen X, et al. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging. 2013;34(6):1564-80.https://doi.org/10.1016/j.neurobiolaging.2012.11.020
- Prajjwal P, Flores Sanga HS, Acharya K, Tango T, John J, Rodriguez RSC, et al. Parkinson's disease updates: Addressing the pathophysiology, risk factors, genetics, diagnosis, along with the medical and surgical treatment. Annals of Medicine & Surgery. 2023;85(10):4887-902.https://doi.org/10.1097/MS9.0000000000001142
- Surmeier DJ. Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J. 2018;285(19):3657-68.https://doi.org/10.1111/febs.14607
- Mehra S, Sahay S, Maji SK. α-Synuclein misfolding and aggregation: Implications in Parkinson's disease pathogenesis. Biochim Biophys Acta Proteins Proteom. 2019;1867(10):890-908.https://doi.org/10.1016/j.bbapap.2019.03.001
- Habibi E, Masoudi-Nejad A, Abdolmaleky HM, Haggarty SJ. Emerging roles of epigenetic mechanisms in Parkinson's disease. Funct Integr Genomics. 2011;11(4):523-37.https://doi.org/10.1007/s10142-011-0246-z
- Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol. 2012;72(6):893-901.https://doi.org/10.1002/ana.23687
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson's Disease. Science. 1997;276(5321):2045-7.https://doi.org/10.1126/science.276.5321.2045
- Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 Mutations in Parkinson Disease. The American Journal of Human Genetics. 2011;89(1):162-7.https://doi.org/10.1016/j.ajhg.2011.06.001
- Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89(3):398-406.https://doi.org/10.1016/j.ajhg.2011.08.009
- Vilariño-Güell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23(7):1794-801.https://doi.org/10.1093/hmg/ddt570
- Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol. 2015;14(3):274-82.https://doi.org/10.1016/S1474-4422(14)70266-2
- Keating DJ. Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem. 2008;104(2):298-305.https://doi.org/10.1111/j.1471-4159.2007.04997.x
- Dantuma NP, Bott LC. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci. 2014;7:70.https://doi.org/10.3389/fnmol.2014.00070
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16(6):345-57.https://doi.org/10.1038/nrn3961
- Sankowski R, Mader S, Valdés-Ferrer SI. Systemic Inflammation and the Brain: Novel Roles of Genetic, Molecular, and Environmental Cues as Drivers of Neurodegeneration. Front Cell Neurosci. 2015;9:28.https://doi.org/10.3389/fncel.2015.00028
- Kalia L V., Lang AE. Parkinson's disease. The Lancet. 2015;386(9996):896-912.https://doi.org/10.1016/S0140-6736(14)61393-3
- Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci. 2006;26(39):9996-10005.https://doi.org/10.1523/JNEUROSCI.3292-06.2006
- Romaní-Aumedes J, Canal M, Martín-Flores N, Sun X, Pérez-Fernández V, Wewering S, et al. Parkin loss of function contributes to RTP801 elevation and neurodegeneration in Parkinson's disease. Cell Death Dis. 2014;5(8):e1364-e1364.https://doi.org/10.1038/cddis.2014.333
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30(3):1166-75.https://doi.org/10.1523/JNEUROSCI.3944-09.2010
- Deguil J, Jailloux D, Page G, Fauconneau B, Houeto JL, Philippe M, et al. Neuroprotective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) in MPP+-induced alteration of translational control in Neuro-2a neuroblastoma cells. J Neurosci Res. 2007;85(9):2017-25.https://doi.org/10.1002/jnr.21318
- Rodríguez‐Blanco J, Martín V, García‐Santos G, Herrera F, Casado‐Zapico S, Antolín I, et al. Cooperative action of JNK and AKT/mTOR in 1‐methyl‐4‐phenylpyridinium‐induced autophagy of neuronal PC12 cells. J Neurosci Res. 2012;90(9):1850-60.https://doi.org/10.1002/jnr.23066
- Deguil J, Chavant F, Lafay-Chebassier C, Pérault-Pochat MC, Fauconneau B, Pain S. Time course of MPTP toxicity on translational control protein expression in mice brain. Toxicol Lett. 2010;196(1):51-5.https://doi.org/10.1016/j.toxlet.2010.03.1121
- Deguil J, Chavant F, Lafay-Chebassier C, Pérault-Pochat MC, Fauconneau B, Pain S. Neuroprotective effect of PACAP on translational control alteration and cognitive decline in MPTP parkinsonian mice. Neurotox Res. 2010;17(2):142-55.https://doi.org/10.1007/s12640-009-9091-4
- Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR Signaling in Parkinson's Disease Prevents L-DOPA-Induced Dyskinesia. Sci Signal. 2009;2(80):ra36.https://doi.org/10.1126/scisignal.2000308
- Zhang H, Duan C, Yang H. Defective Autophagy in Parkinson's Disease: Lessons from Genetics. Mol Neurobiol. 2015;51(1):89-104.https://doi.org/10.1007/s12035-014-8787-5
- Cartier AE, Ubhi K, Spencer B, Vazquez-Roque RA, Kosberg KA, Fourgeaud L, et al. Differential effects of UCHL1 modulation on alpha-synuclein in PD-like models of alpha-synucleinopathy. PLoS One. 2012;7(4):e34713.https://doi.org/10.1371/journal.pone.0034713
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394-406.https://doi.org/10.1038/nn.3350
- Vives-Bauza C, Zhou C, Huang Y, Cui M, De Vries RLA, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378-83.https://doi.org/10.1073/pnas.0911187107
- Rakovic A, Grünewald A, Kottwitz J, Brüggemann N, Pramstaller PP, Lohmann K, et al. Mutations in PINK1 and Parkin Impair Ubiquitination of Mitofusins in Human Fibroblasts. PLoS One. 2011;6(3):e16746.https://doi.org/10.1371/journal.pone.0016746
- Trempe J-F, Fon EA. Structure and Function of Parkin, PINK1, and DJ-1, the Three Musketeers of Neuroprotection. Front Neurol. 2013;4:38.https://doi.org/10.3389/fneur.2013.00038
- Son SM, Siddiqi FH, Lopez A, Ansari R, Tyrkalska SD, Park SJ, et al. Alpha-synuclein mutations mislocalize cytoplasmic p300 compromising autophagy, which is rescued by ACLY inhibition. Neuron. 2025;113(12):1908-1924.e13.https://doi.org/10.1016/j.neuron.2025.03.028
- Jiang TF, Zhang YJ, Zhou HY, Wang HM, Tian LP, Liu J, et al. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson's disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. Journal of Neuroimmune Pharmacology. 2013;8(1):356-69.https://doi.org/10.1007/s11481-012-9431-7
- Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces α-synuclein aggregation and toxicity. Journal of Biological Chemistry. 2004;279(24):25497-502.https://doi.org/10.1074/jbc.M400255200
- Poehler A-M, Xiang W, Spitzer P, May VEL, Meixner H, Rockenstein E, et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy. 2014;10(12):2171-92.https://doi.org/10.4161/auto.36436
- Testa CM, Jankovic J. Huntington disease: A quarter century of progress since the gene discovery. J Neurol Sci. 2019;396:52-68.https://doi.org/10.1016/j.jns.2018.09.022
- Walker FO. Huntington's disease. The Lancet. 2007;369(9557):218-28.https://doi.org/10.1016/S0140-6736(07)60111-1
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90(3):905-81.https://doi.org/10.1152/physrev.00041.2009
- Thompson PD, Berardelli A, Rothwell JC, Day BL, Dick JPR, Benecke R, et al. The coexistence of bradykinesia and chorea in huntington's disease and its implications for theories of basal ganglia control of movement. Brain. 1988;111(2):223-44.https://doi.org/10.1093/brain/111.2.223
- Stout JC, Paulsen JS, Queller S, Solomon AC, Whitlock KB, Campbell JC, et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology. 2011;25(1):1-14.https://doi.org/10.1037/a0020937
- Taherzadeh-Fard E, Saft C, Akkad DA, Wieczorek S, Haghikia A, Chan A, et al. PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of Huntington disease. Mol Neurodegener. 2011;6(1):32.https://doi.org/10.1186/1750-1326-6-32
- Saft C, Epplen JT, Wieczorek S, Landwehrmeyer GB, Roos RAC, de Yebenes JG, et al. NMDA receptor gene variations as modifiers in Huntington disease: a replication study. PLoS Curr. 2011;3:RRN1247.https://doi.org/10.1371/currents.RRN1247
- Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, et al. Cerebral cortex and the clinical expression of Huntington's disease: complexity and heterogeneity. Brain. 2008;131(4):1057-68.https://doi.org/10.1093/brain/awn025
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, et al. Evidence for more widespread cerebral pathology in early HD. Neurology. 2003;60(10):1615-20.https://doi.org/10.1212/01.WNL.0000065888.88988.6E
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24(17):4250-8.https://doi.org/10.1523/JNEUROSCI.3920-03.2004
- Lee JH, Tecedor L, Chen YH, Monteys AM, Sowada MJ, Thompson LM, et al. Reinstating aberrant mTORC1 activity in huntington's disease mice improves disease phenotypes. Neuron. 2015;85(2):303-15.https://doi.org/10.1016/j.neuron.2014.12.019
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585-95.https://doi.org/10.1038/ng1362
- Pryor WM, Biagioli M, Shahani N, Swarnkar S, Huang W-C, Page DT, et al. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington's disease. Sci Signal. 2014;7(349):ra103.https://doi.org/10.1126/scisignal.2005633
- Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH. Rhes, a Striatal-selective Protein Implicated in Huntington Disease, Binds Beclin-1 and activates autophagy. Journal of Biological Chemistry. 2014;289(6):3547-54.https://doi.org/10.1074/jbc.M113.536912
- Rui Y-N, Xu Z, Patel B, Chen Z, Chen D, Tito A, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17(3):262-75.https://doi.org/10.1038/ncb3101
- Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. Journal of Biological Chemistry. 2009;284(18):12297-305.https://doi.org/10.1074/jbc.M900573200
- Riva N, Domi T, Pozzi L, Lunetta C, Schito P, Spinelli EG, et al. Update on recent advances in amyotrophic lateral sclerosis. J Neurol. 2024;271(7):4693-723.https://doi.org/10.1007/s00415-024-12435-9
- Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, et al. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6(1):171.https://doi.org/10.4103/2152-7806.169561
- Benatar M, Robertson J, Andersen PM. Amyotrophic lateral sclerosis caused by SOD1 variants: from genetic discovery to disease prevention. Lancet Neurol. 2025;24(1):77-86.https://doi.org/10.1016/S1474-4422(24)00479-4
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396-403.https://doi.org/10.1038/nn.2660
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien J-P. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9(1):108-18.https://doi.org/10.1038/nn1603
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science. 2008;319(5870):1668-72.https://doi.org/10.1126/science.1154584
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science. 2009;323(5918):1208-11.https://doi.org/10.1126/science.1165942
- Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M, et al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J Neuroimmunol. 2009;210(1-2):73-9.https://doi.org/10.1016/j.jneuroim.2009.02.012
- Martin LJ. Mitochondrial pathobiology in ALS. J Bioenerg Biomembr. 2011 Dec;43(6):569-79.https://doi.org/10.1007/s10863-011-9395-y
- Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7(4):412-25.https://doi.org/10.4161/auto.7.4.14541
- Carunchio I, Curcio L, Pieri M, Pica F, Caioli S, Viscomi MT, et al. Increased levels of p70S6 phosphorylation in the G93A mouse model of Amyotrophic Lateral Sclerosis and in valine-exposed cortical neurons in culture. Exp Neurol. 2010;226(1):218-30.https://doi.org/10.1016/j.expneurol.2010.08.033
- Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15(7):1209-16.https://doi.org/10.1093/hmg/ddl036
- Barnes PJ. Mechanisms of development of multimorbidity in the elderly. European Respiratory Journal. 2015;45(3):790-806.https://doi.org/10.1183/09031936.00229714
- Schreiber KH, Arriola Apelo SI, Yu D, Brinkman JA, Velarde MC, Syed FA, et al. A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat Commun. 2019;10(1):3194.https://doi.org/10.1038/s41467-019-11174-0
- Kang SA, O'Neill DJ, Machl AW, Lumpkin CJ, Galda SN, Sengupta S, et al. Discovery of Small-Molecule Selective mTORC1 Inhibitors via Direct Inhibition of Glucose Transporters. Cell Chem Biol. 2019;26(9):1203-1213.e13.https://doi.org/10.1016/j.chembiol.2019.05.009
- Luthra R, Roy A. Role of Medicinal Plants against Neurodegenerative Diseases. Curr Pharm Biotechnol. 2022;23(1):123-39.https://doi.org/10.2174/1389201022666210211123539
- Ratheesh G, Tian L, Venugopal JR, Ezhilarasu H, Sadiq A, Fan T-P, et al. Role of medicinal plants in neurodegenerative diseases. Biomanufacturing Reviews. 2017;2(1):2.https://doi.org/10.1007/s40898-017-0004-7
- Hu K, Wu S, Xu J, Zhang Y, Zhang Y, Wu X, et al. Pongamol Alleviates Neuroinflammation and Promotes Autophagy in Alzheimer's Disease by Regulating the Akt/mTOR Signaling Pathway. J Agric Food Chem. 2024;72(24):13634-45.https://doi.org/10.1021/acs.jafc.4c00836
- Chen M, Peng L, Gong P, Zheng X, Sun T, Zhang X, et al. Baicalein Induces Mitochondrial Autophagy to Prevent Parkinson's Disease in Rats via miR-30b and the SIRT1/AMPK/mTOR Pathway. Front Neurol. 2022;12:643817.https://doi.org/10.3389/fneur.2021.646817
- Liu Z, Zhuang W, Cai M, Lv E, Wang Y, Wu Z, et al. Kaemperfol Protects Dopaminergic Neurons by Promoting mTOR-Mediated Autophagy in Parkinson's Disease Models. Neurochem Res. 2023;48(5):1395-411.https://doi.org/10.1007/s11064-022-03819-2
- Van Skike CE, Hussong SA, Hernandez SF, Banh AQ, DeRosa N, Galvan V. mTOR Attenuation with Rapamycin Reverses Neurovascular Uncoupling and Memory Deficits in Mice Modeling Alzheimer's Disease. The Journal of Neuroscience. 2021;41(19):4305-20.https://doi.org/10.1523/JNEUROSCI.2144-20.2021
- Yu L, Hu X, Xu R, Zhao Y, Xiong L, Ai J, et al. Piperine promotes PI3K/AKT/mTOR-mediated gut-brain autophagy to degrade α-Synuclein in Parkinson's disease rats. J Ethnopharmacol. 2024;322:117628.https://doi.org/10.1016/j.jep.2023.117628
- Zhu Z, Yang C, Iyaswamy A, Krishnamoorthi S, Sreenivasmurthy SG, Liu J, et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson's Disease. Int J Mol Sci. 2019;20(3):728.https://doi.org/10.3390/ijms20030728
- St-Cyr S, Child DD, Giaime E, Smith AR, Pascua CJ, Hahm S, et al. Huntington's disease phenotypes are improved via mTORC1 modulation by small molecule therapy. PLoS One. 2022;17(8):e0273710.https://doi.org/10.1371/journal.pone.0273710
- Granatiero V, Sayles NM, Savino AM, Konrad C, Kharas MG, Kawamata H, et al. Modulation of the IGF1R-MTOR pathway attenuates motor neuron toxicity of human ALS SOD1 G93A astrocytes. Autophagy. 2021;17(12):4029-42.https://doi.org/10.1080/15548627.2021.1899682
- Van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari Z, et al. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer's disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol. 2018;314(4):H693-703.https://doi.org/10.1152/ajpheart.00570.2017
- Wang X, Jia J. Magnolol improves Alzheimer's disease-like pathologies and cognitive decline by promoting autophagy through activation of the AMPK/mTOR/ULK1 pathway. Biomed. Pharmacother. 2023;161:114473.https://doi.org/10.1016/j.biopha.2023.114473
- Cheng X, Dai Y, Shang B, Zhang S, Lin L, Wu Q, et al. Danggui Shaoyao San and disassembled prescription: neuroprotective effects via AMPK/mTOR-mediated autophagy in mice. BMC Complement Altern Med. 2024;24(1):1-16.https://doi.org/10.1186/s12906-024-04588-x
- Salem MA, Budzyńska B, Kowalczyk J, El Sayed NS, Mansour SM. Tadalafil and bergapten mitigate streptozotocin-induced sporadic Alzheimer's disease in mice via modulating neuroinflammation, PI3K/Akt, Wnt/β-catenin, AMPK/mTOR signaling pathways. Toxicol Appl Pharmacol. 2021;429:115697.https://doi.org/10.1016/j.taap.2021.115697
- Wu X, Su D, Xu J, Ge G, Zhang Y, Wu B, et al. Tricetin, a Dietary Flavonoid, Alleviates Neuroinflammation and Promotes Autophagy in Alzheimer's Disease by Regulating the PI3K/Akt/mTOR Signaling Pathway. J Agric Food Chem. 2025;73(16):9677-89.https://doi.org/10.1021/acs.jafc.5c01158
- Mao J, Cheng L, Zhang Y dan, Xie G jing, Wang P. Chinese formula Guben-Jiannao Ye alleviates the dysfunction of circadian and sleep rhythms in APP/PS1 mice implicated in activation of the PI3K/AKT/mTOR signaling pathway. J Ethnopharmacol. 2024;335:118696.https://doi.org/10.1016/j.jep.2024.118696
- Zhu Z, Liu Y, Li X, Zhang L, Liu H, Cui Y, et al. GPNMB mitigates Alzheimer's disease and enhances autophagy via suppressing the mTOR signal. Neurosci Lett. 2022;767:136300.https://doi.org/10.1016/j.neulet.2021.136300
- Wang L, Tian S, Ruan S, Wei J, Wei S, Chen W, et al. Neuroprotective effects of cordycepin on MPTP-induced Parkinson's disease mice via suppressing PI3K/AKT/mTOR and MAPK-mediated neuroinflammation. Free Radic Biol Med. 2024;216:60-77.https://doi.org/10.1016/j.freeradbiomed.2024.02.023
- Pan Y, Chen M, Pan L, Tong Q, Cheng Z, Lin S, et al. Shisandra Decoction Alleviates Parkinson's Disease Symptoms in a Mouse Model Through PI3K/AKT/mTOR Signalling Pathway. Neuropsychiatr Dis Treat. 2024;20:2011-27.https://doi.org/10.2147/NDT.S476969
- El-Sayed RM, Abdelaziz AM, Zaki HF, Abdel Rasheed NO. Cilostazol novel neuroprotective mechanism against rotenone-induced Parkinson's disease in rats: Correlation between Nrf2 and HMGB1/TLR4/PI3K/Akt/mTOR signaling. Int Immunopharmacol. 2023;117:109986.https://doi.org/10.1016/j.intimp.2023.109986
- Zhao N, Wu M, Velu P, Annamalai V, Zhang J. Sanggenol L Alleviates Rotenone-induced Parkinson's Disease and Inhibits Mitochondrial Complex I by Apoptosis Via P13K/AKT/mTOR Signalling. Comb Chem High Throughput Screen. 2024;28. DOI: 10.2174/0113862073358649241128053921https://doi.org/10.2174/0113862073358649241128053921
- Zhai H, Kang Z, Zhang H, Ma J, Chen G. Baicalin attenuated substantia nigra neuronal apoptosis in Parkinson's disease rats via the mTOR/AKT/GSK-3β pathway. J Integr Neurosci. 2019;18(4):423-9.https://doi.org/10.31083/j.jin.2019.04.192
- Park JS, Choe K, Lee HJ, Park TJ, Kim MO. Neuroprotective effects of osmotin in Parkinson's disease-associated pathology via the AdipoR1/MAPK/AMPK/mTOR signaling pathways. J Biomed Sci. 2023;30(1):66.https://doi.org/10.1186/s12929-023-00961-z
- Shirgadwar SM, Kumar R, Preeti K, Khatri DK, Singh SB. Neuroprotective Effect of Phloretin in Rotenone-Induced Mice Model of Parkinson's Disease: Modulating mTOR-NRF2-p62 Mediated Autophagy-Oxidative Stress Crosstalk. Journal of Alzheimer's Disease. 2023;94(s1):S109-24.https://doi.org/10.3233/JAD-220793
- Zhou Q, Chen B, Xu Y, Wang Y, He Z, Cai X, et al. Geniposide protects against neurotoxicity in mouse models of rotenone-induced Parkinson's disease involving the mTOR and Nrf2 pathways. J Ethnopharmacol. 2024;318(Pt A):116914.https://doi.org/10.1016/j.jep.2023.116914
- Salama RM, Abdel-Latif GA, Abbas SS, El Magdoub HM, Schaalan MF. Neuroprotective effect of crocin against rotenone-induced Parkinson's disease in rats: Interplay between PI3K/Akt/mTOR signaling pathway and enhanced expression of miRNA-7 and miRNA-221. Neuropharmacology. 2020;164:107900.https://doi.org/10.1016/j.neuropharm.2019.107900
- Qi H, Shen D, Jiang C, Wang H, Chang M. Ursodeoxycholic acid protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis in MPTP/MPP+-induced Parkinson's disease. Neurosci Lett. 2021;741:135493.https://doi.org/10.1016/j.neulet.2020.135493
- Nataraj J, Manivasagam T, Justin Thenmozhi A, Essa MM. Neurotrophic Effect of Asiatic acid, a Triterpene of Centella asiatica Against Chronic 1-Methyl 4-Phenyl 1, 2, 3, 6-Tetrahydropyridine Hydrochloride/Probenecid Mouse Model of Parkinson's disease: The Role of MAPK, PI3K-Akt-GSK3β and mTOR Signalling Pathways. Neurochem Res. 2017;42(5):1354-65.https://doi.org/10.1007/s11064-017-2183-2
- Zhang L, Park JY, Zhao D, Kwon HC, Yang HO. Neuroprotective Effect of Astersaponin I against Parkinson's Disease through Autophagy Induction. Biomol Ther. 2021;29(6):615-29.https://doi.org/10.4062/biomolther.2021.004
- Ni C, Wang L, Bai Y, Huang F, Shi H, Wu H, et al. Taurochenodeoxycholic acid activates autophagy and suppresses inflammatory responses in microglia of MPTP-induced Parkinson's disease mice via AMPK/mTOR, AKT/NFκB and Pink1/Parkin signaling pathways mediated by Takeda G protein-coupled receptor 5. Free Radic Biol Med. 2025;235:347-63.https://doi.org/10.1016/j.freeradbiomed.2025.04.053
- Wang X, Pang L, Zhang Y, Xu J, Ding D, Yang T, et al. Lycium barbarum Polysaccharide Promotes Nigrostriatal Dopamine Function by Modulating PTEN/AKT/mTOR Pathway in a Methyl-4-phenyl-1, 2,3, 6-tetrahydropyridine (MPTP) Murine Model of Parkinson's Disease. Neurochem Res. 2018;43(4):938-47.https://doi.org/10.1007/s11064-018-2499-6
- Gugliandolo A, Pollastro F, Bramanti P, Mazzon E. Cannabidiol exerts protective effects in an in vitro model of Parkinson's disease activating AKT/mTOR pathway. Fitoterapia. 2020;143:104553.https://doi.org/10.1016/j.fitote.2020.104553
- He PK, Gao YY, Lyu F-J, Chen JN, Zhang YH, Nie K, et al. Idebenone-Activating Autophagic Degradation of α-Synuclein via Inhibition of AKT-mTOR Pathway in a SH-SY5Y-A53T Model of Parkinson's Disease: A Network Pharmacological Approach. Evidence-Based Complementary and Alternative Medicine. 2021;2021:1-14.https://doi.org/10.1155/2021/8548380
- Jiang D, Peng Y. The protective effect of decoction of Rehmanniae via PI3K/Akt/mTOR pathway in MPP+-induced Parkinson's disease model cells. J Recept Signal Transduct Res. 2021;41(1):74-84.https://doi.org/10.1080/10799893.2020.1787445
- Yang S, Wan Y, Wu N, Song L, Liu Z, Zhao J, et al. L-3, 4-Dihydroxyphenylalanine Recovers Circadian Rhythm Disturbances in the Rat Models of Parkinson's Disease by Regulating the D1R-ERK1/2-mTOR Pathway. Front Aging Neurosci. 2021;13:719885.https://doi.org/10.3389/fnagi.2021.719885
- Choi Y, Huh E, Lee S, Kim JH, Park MG, Seo SY, et al. 5-Hydroxytryptophan Reduces Levodopa-Induced Dyskinesia via Regulating AKT/mTOR/S6K and CREB/ΔFosB Signals in a Mouse Model of Parkinson's Disease. Biomol Ther. 2023;31(4):402-10.https://doi.org/10.4062/biomolther.2022.141
- Shi L, Huang C, Luo Q, Xia Y, Liu W, Zeng W, et al. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson's disease through AKT/mTOR pathway. Aging. 2020;12(10):9515-33.https://doi.org/10.18632/aging.103225
- El-Emam MA, Sheta E, El-Abhar HS, Abdallah DM, El Kerdawy AM, Eldehna WM, et al. Morin suppresses mTORc1/IRE-1α/JNK and IP3R-VDAC-1 pathways: Crucial mechanisms in apoptosis and mitophagy inhibition in experimental Huntington's disease, supported by in silico molecular docking simulations. Life Sci. 2024;338:122362.https://doi.org/10.1016/j.lfs.2023.122362
- Senousy MA, Hanafy ME, Shehata N, Rizk SM. Erythropoietin and Bacillus Calmette-Guérin Vaccination Mitigate 3-Nitropropionic Acid-Induced Huntington-like Disease in Rats by Modulating the PI3K/Akt/mTOR/P70S6K Pathway and Enhancing the Autophagy. ACS Chem Neurosci. 2022;13(6):721-32.https://doi.org/10.1021/acschemneuro.1c00523
- Abd-Elrahman KS, Ferguson SSG. Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington's pathology in zQ175 mice. Mol Brain. 2019;12(1):35.https://doi.org/10.1186/s13041-019-0456-1
- Saad MA, Ahmed MAE, Elbadawy NN, Abdelkader NF. Nano-ivabradine averts behavioral anomalies in Huntington's disease rat model via modulating Rhes/m-tor pathway. Prog Neuropsychopharmacol Biol Psychiatry. 2021;111:110368.https://doi.org/10.1016/j.pnpbp.2021.110368
- Milani M, Della Valle I, Rossi S, Fabbrizio P, Margotta C, Nardo G, et al. Neuroprotective effects of niclosamide on disease progression via inflammatory pathways modulation in SOD1-G93A and FUS-associated amyotrophic lateral sclerosis models. Neurotherapeutics. 2024;21(3):e00346.https://doi.org/10.1016/j.neurot.2024.e00346
- Sharma R, Mehan S, Khan Z, Das Gupta G, Narula AS. Therapeutic potential of oleanolic acid in modulation of PI3K/Akt/mTOR/STAT-3/GSK-3β signaling pathways and neuroprotection against methylmercury-induced neurodegeneration. Neurochem Int. 2024;180:105876.https://doi.org/10.1016/j.neuint.2024.105876
- Zheng C, Li W, Ali T, Peng Z, Liu J, Pan Z, et al. Ibrutinib Delays ALS Installation and Increases Survival of SOD1G93A Mice by Modulating PI3K/mTOR/Akt Signaling. Journal of Neuroimmune Pharmacology. 2023;18(3):383-96.https://doi.org/10.1007/s11481-023-10068-9
- Kumar S, Mehan S, Khan Z, Das Gupta G, Narula AS. Guggulsterone Selectively Modulates STAT-3, mTOR, and PPAR-Gamma Signaling in a Methylmercury-Exposed Experimental Neurotoxicity: Evidence from CSF, Blood Plasma, and Brain Samples. Mol Neurobiol. 2024;61(8):5161-93.https://doi.org/10.1007/s12035-023-03902-x
- Yang YH, Hsieh SW, Chang HW, Sung JL, Chuu CP, Yen CW, et al. Gamma Frequency Inhibits the Secretion and Aggregation of Amyloid-β and Decreases the Phosphorylation of mTOR and Tau Proteins in vitro. Journal of Alzheimer's Disease. 2022;90(2):917-28.https://doi.org/10.3233/JAD-220307
- Shi Q, Chang C, Saliba A, Bhat MA. Microglial mTOR Activation Upregulates Trem2 and Enhances β-Amyloid Plaque Clearance in the 5XFAD Alzheimer's Disease Model. The Journal of Neuroscience. 2022;42(27):5294-313.https://doi.org/10.1523/JNEUROSCI.2427-21.2022
- Cheng L, Chen Y, Guo D, Zhong Y, Li W, Lin Y, et al. mTOR-dependent TFEB activation and TFEB overexpression enhance autophagy-lysosome pathway and ameliorate Alzheimer's disease-like pathology in diabetic encephalopathy. Cell Commun Signal. 2023;21(1):91.https://doi.org/10.1186/s12964-023-01097-1
- Azimzadeh M, Cheah PS, Ling KH. Brain insulin resistance in Down syndrome: Involvement of PI3K-Akt/mTOR axis in early-onset of Alzheimer's disease and its potential as a therapeutic target. Biochem Biophys Res Commun. 2024;733:150713.https://doi.org/10.1016/j.bbrc.2024.150713
- Yun Q, Ma SF, Zhang WN, Gu M, Wang J. FoxG1 as a Potential Therapeutic Target for Alzheimer's Disease: Modulating NLRP3 Inflammasome via AMPK/mTOR Autophagy Pathway. Cell Mol Neurobiol. 2024;44(1):35.https://doi.org/10.1007/s10571-024-01467-4
- Franco R, Martínez-Pinilla E, Navarro G, Zamarbide M. Potential of GPCRs to modulate MAPK and mTOR pathways in Alzheimer's disease. Prog Neurobiol. 2017;149-150:21-38.https://doi.org/10.1016/j.pneurobio.2017.01.004
- Ebrahim N, Al Saihati HA, Alali Z, Aleniz FQ, Mahmoud SYM, Badr OA, et al. Exploring the molecular mechanisms of MSC-derived exosomes in Alzheimer's disease: Autophagy, insulin and the PI3K/Akt/mTOR signaling pathway. Biomed Pharmacother. 2024;176:116836.https://doi.org/10.1016/j.biopha.2024.116836
- Li Y, Pang J, Wang J, Dai G, Bo Q, Wang X, et al. Knockdown of PDCD4 ameliorates neural cell apoptosis and mitochondrial injury through activating the PI3K/AKT/mTOR signal in Parkinson's disease. J Chem Neuroanat. 2023;129:102239.https://doi.org/10.1016/j.jchemneu.2023.102239
- Zhang Z, Sun X, Wang K, Yu Y, Zhang L, Zhang K, et al. Hydrogen-saturated saline mediated neuroprotection through autophagy via PI3K/AKT/mTOR pathway in early and medium stages of rotenone-induced Parkinson's disease rats. Brain Res Bull. 2021;172:1-13.https://doi.org/10.1016/j.brainresbull.2021.04.003
- Qian C, Ye Y, Mao H, Yao L, Sun X, Wang B, et al. Downregulated lncRNA-SNHG1 enhances autophagy and prevents cell death through the miR-221/222 /p27/mTOR pathway in Parkinson's disease. Exp Cell Res. 2019;384(1):111614.https://doi.org/10.1016/j.yexcr.2019.111614
- Ba RQ, Liu J, Fan XJ, Jin GL, Huang BG, Liu MW, et al. Effects of miR-199a on autophagy by targeting glycogen synthase kinase 3β to activate PTEN/AKT/mTOR signaling in an MPP+ in vitro model of Parkinson's disease. Neurol Res. 2020;42(4):308-18.https://doi.org/10.1080/01616412.2020.1726584
- Ge H, Yan Z, Zhu H, Zhao H. MiR-410 exerts neuroprotective effects in a cellular model of Parkinson's disease induced by 6-hydroxydopamine via inhibiting the PTEN/AKT/mTOR signaling pathway. Exp Mol Pathol. 2019;109:16-24.https://doi.org/10.1016/j.yexmp.2019.05.002
- Li W, Jiang Y, Wang Y, Yang S, Bi X, Pan X, et al. MiR-181b regulates autophagy in a model of Parkinson's disease by targeting the PTEN/Akt/mTOR signaling pathway. Neurosci Lett. 2018;675:83-8.https://doi.org/10.1016/j.neulet.2018.03.041
- Wen Z, Zhang J, Tang P, Tu N, Wang K, Wu G. Overexpression of miR 185 inhibits autophagy and apoptosis of dopaminergic neurons by regulating the AMPK/mTOR signaling pathway in Parkinson's disease. Mol Med Rep. 2017;17(1):131-7.https://doi.org/10.3892/mmr.2017.7897
- Gong X, Wang H, Ye Y, Shu Y, Deng Y, He X, et al. miR-124 regulates cell apoptosis and autophagy in dopaminergic neurons and protects them by regulating AMPK/mTOR pathway in Parkinson's disease. Am J Transl Res. 2016;8(5):2127-37.
- Wang S, Wang Q, Ma J, Yu P, Wang Z, Wang B. Effect of moxibustion on mTOR-mediated autophagy in rotenone-induced Parkinson's disease model rats. Neural Regen Res. 2018;13(1):112.https://doi.org/10.4103/1673-5374.224380
- Ning B, Wang Z, Wu Q, Deng Q, Yang Q, Gao J, et al. Acupuncture inhibits autophagy and repairs synapses by activating the mTOR pathway in Parkinson's disease depression model rats. Brain Res. 2023;1808:148320. https://doi.org/10.1016/j.brainres.2023.148320
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295-305. https://doi.org/10.1038/nchembio.79
- Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum Mol Genet. 2008;17(2):170-8. https://doi.org/10.1093/hmg/ddm294
- Li C, Chen Y, Chen X, Wei Q, Cao B, Shang H. Downregulation of MicroRNA-193b-3p Promotes Autophagy and Cell Survival by Targeting TSC1/mTOR Signaling in NSC-34 Cells. Front Mol Neurosci. 2017;10:160. https://doi.org/10.3389/fnmol.2017.00160