Original Article – DOI: 10.33594/000000869
CPB (60): 260 - 269
Accepted: 29.04.2026 - Published: 11.05.2026
Effects of Urolithin A on Mitochondrial Homeostasis Disruption by LPS in C2C12 Myotubes
bInstitute of Neuroscience, UCLouvain, Louvain-la-Neuve, Belgium,
cCentro de Salud Deportiva. Clínica Santa María, Santiago, Chile,
dInstitute of Health and Sport Sciences, Faculty of Health Science, Universidad Francisco de Vitoria, Madrid, Spain.,
eExercise and Rehabilitation Science Institute, Faculty of Rehabilitation Sciences, School of physical therapy, Universidad Andres Bello, Santiago, Chile,
fCentro de salud y ejercicio Suizmed, Santiago, Chile
Keywords
Abstract
Background/Aims: The Toll-like receptor 4 (TLR4) pathway plays a critical role in mediating inflammatory responses and regulates mitochondrial structural adaptations. By inducing mito- and autophagy, urolithin A (UA), a recent natural compound, may potentially enhance mitochondrial homeostasis and health. The aim of this study was to examine the effects of UA on key components and regulators of TLR4 signaling, mitochondrial dynamics and autophagy, and to analyze mitochondrial morphology after lipopolysaccharide (LPS) incubation in C2C12 myotubes. Methods: On day 6 of differentiation, C2C12 myotubes were incubated with LPS (1µg/ml) for 3h and/or UA (50µM) for 24h. Protein expressions associated with TLR4 signaling, mitochondrial dynamics and autophagy were determined by Western blot, and mitochondrial morphology was assessed using electron microscopy. Results: 1) LPS-induced inflammation activated downstream TLR4 signaling, by increasing the expression of inhibitor of nuclear factor-κB (IκBα, p=0.05) and the phosphorylation of extracellular signal-regulated kinase (ERK, p=0.003), p38 (p=0.04) and c-Jun NH(2)-terminal kinase (JNK, p=0.006) and tended to increase the phosphorylation of phospho-transforming growth factor-β-activated kinase 1 (TAK1, p=0.06) and phospho-inhibitor of nuclear factor-κB kinase α/β (IKKαb, p=0.07); 2) LPS reduced mitochondrial area (p=0.05) and circularity (p=0.0004) and tended to reduce mitochondrial perimeter (p=0.07); 3) LPS increased the phosphorylation of the fission marker dynamin-related protein 1 (DRP1, p=0.009); 4) UA completely prevented, or at least attenuated, all previous effects induced by LPS. Conclusion: Our findings suggest that UA partially mitigates LPS-induced inflammation by modulating the TLR4 signaling pathway and mitochondrial dynamics. Even though no causal relation may be established based on the present findings, UA may represent a promising therapeutic agent for conditions associated with mitochondrial dysfunction and inflammation, highlighting its potential role in promoting skeletal muscle health.
Introduction
Urolithin A (UA) is a gut microbiota-derived metabolite that is produced upon the breakdown of ellagitannins - polyphenols found in foods such as pomegranates and berries (1). This compound has gained significant attention in recent years due to its potential health benefits, particularly for muscle function and mitochondrial health (2). UA has been shown to stimulate mitophagy, a cellular process that removes dysfunctional mitochondria through mitophagolysosomes, mediated by proteins involved in both canonical and non-canonical pathways (3), thereby stimulating mitochondrial biogenesis to maintain cellular mitochondrial homeostasis and respiratory capacity (4). Mitophagy is regulated by both canonical and non-canonical pathways, involving proteins such as phosphatase and tensin homolog-induced putative kinase 1 (PINK1), Parkin, and Bcl-2 interacting protein 3 (BNIP3) (3). One recent study has reported that UA can restore mitochondrial and muscle function in murine models of muscular dystrophy, where mitochondrial dysfunction is a hallmark feature (5). Mitochondria are highly dynamic organelles that undergo constant fusion and fission processes. These processes are essential for maintaining mitochondrial health and function. Fusion allows for the exchange of mitochondrial contents and can help mitigate damage, while fission is important for the segregation of damaged mitochondria for subsequent degradation through mitophagy (6). The balance between mitochondrial fusion and fission is essential for cellular homeostasis, and its disruption has been implicated in various pathological conditions, including myopathies (7). Dynamin-related protein 1 (DRP1) is a key regulator of mitochondrial fission. The phosphorylation of DRP1 (p-DRP1) plays a crucial role in its activation and recruitment to the mitochondrial outer membrane, where it facilitates fission (8). Interestingly, recent studies have shown that mitophagy can be associated with p-DRP1 regardless of mitochondrial morphology (9), suggesting a complex interaction between mitochondrial dynamics and quality control mechanisms. Mitochondrial damage is sensed by the inflammasome, which triggers IL-1β production and the recruitment of the innate immune system to the affected tissues (10). Also, a damage-associated molecular pattern (DAMP) can enter the circulation, where it binds to and activates pattern recognition receptors (PRRs), such as Toll-like receptor 4 (TLR4) (11). Mitophagy inhibition elevates reactive oxygen species (ROS) production and inflammasome activity. TLR4 signalling triggers the activation of the nuclear factor-κ B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, leading to the production of pro-inflammatory cytokines such as tumoral nuclear factor alpha (TNFα), interleukin 1 (IL-1), interleukin-6 (IL-6), interleukin-12 (IL-12) and type-I interferons to mount a strong inflammatory response (11). Lipopolysaccharide (LPS), a component of gram-negative bacterial cell walls, is a potent activator of TLR4 signaling. Recent studies have shown that LPS-induced inflammation can affect mitochondrial morphology in human myotubes, highlighting the interconnection between inflammation and mitochondrial dysfunction in muscle cells (12). UA has been found to have anti-inflammatory effects in various disease models via, amongst others, the inhibition of the NF-κB pathway (13, 14). This suggests that UA may have a dual role in protecting muscle cells, namely by stimulating mitophagy and by mitigating inflammation. Given the complex interplay between inflammation, mitochondrial dysfunction, and muscle health, we hypothesized that UA could reduce the activation of components of the TLR4 inflammatory pathway following acute LPS stimulation. Furthermore, UA could restore optimal mitochondrial morphology in C2C12 myotubes, potentially through its effects on mitophagy. To investigate this, we examined the effects of UA on key components and regulators of TLR4 signaling, mitochondrial dynamics and autophagy, and analyzed mitochondrial morphology.
Materials and Methods
Cell culture
C2C12 murine skeletal muscle myoblasts (ATCC) were maintained in complete DMEM (Dulbecco’s modifed Eagle’s medium) supplemented with 10% FBS and 1% antibiotic/antimycotic and incubated at 37°C and 5% CO2. Once at 80% confluence, the culture medium was changed to 1% horse serum to induce the differentiation process.
Experimental procedures
On the sixth day of differentiation, the cells were treated with 50µM UA (3, 8-dihydroxy-6H-dibenzo [b,d]pyran-6-one) for 24h, using DMSO as the vehicle, as previously described (15). Cells were treated with LPS at 1µg/ml for 3h (PBS as the vehicle) from 21h with UA. The four experimental conditions were: 1) CTL, 2) LPS, 3) UA and 4) UA +LPS.
Western blotting
C2C12 myotubes were homogenized in a lysis buffer containing: 20 mM Tris-HCl (pH 7.5), 1% Triton X‐100, 2 mM EDTA, 20 mM NaF, 1 mM Na2P2O7, 10% glycerol, 150 mM NaCl, 10 mM Na3VO4, 1 mM PMSF, and a protease inhibitor cocktail (Complete TM, Roche Applied Science). Fifteen µg of protein were loaded per sample, separated by SDS-PAGE and transfered to a PVDF membrane with a pore size of 0.2µm (1620177, BIO-RAD). The following antibodies were used: TLR4 (sc-293072, Santa-Cruz Biotechnology), myeloid differentiation primary response 88 (MyD88) (sc-74532, Santa-Cruz Biotechnology), phospho-transforming growth factor-β-activated kinase 1 (TAK1) (Ser412) (9339S, Cell Signaling), phospho-inhibitor of nuclear factor-κB kinase α/β (IKKα/β) (Ser176/180) (2697S, Cell Signaling), inhibitor of nuclear factor-κB kinase α (IκBα) (4814S, Cell Signaling), p65 NF-κB (8242S, Cell Signaling), phospho-stress-activated protein kinase/c-Jun NH(2)-terminal kinase (SAPK/JNK) (Thr183/Tyr185) (9251S, Cell Signaling), phospho-p38 MAPK (Thr180/Tyr182) (9215S, Cell Signaling), phospho-p44/42 extracellular signal-regulated kinase (ERK1/2) MAPK (Thr202/Tyr204) (4695S, Cell Signaling), phospho-DRP1 (Ser616) (4494S, Cell Signaling), optic atrophy 1 (Opa1) (612606, BD Biosciences), PINK1 (BC100-494SS, Novus Biologicals), phospho-adenosine monophosphate-activated protein kinase (AMPK) (Thr172) (2531S, Cell Signaling), p62 (5114S, Cell Signaling), phospho-Unc-51-like kinase (ULK) (Ser757) (14202S, Cell Signaling), microtubule-associated protein 1A/1B-light chain 3 (LC3I-II) (4108S, Cell Signaling). Subsequently, the membranes were incubated with the corresponding secondary antibody, goat anti-rabbit IgG-horseradish peroxidase (7074P2, Cell Signaling) and goat anti-mouse IgG-horseradish peroxidase (31430, Invitrogen). The ChemiDoc Imaging System was used to obtain images that were subsequently analyzed using the Image Lab software (Bio-Rad). Protein expressions were normalized to Ponceau S staining (BM-1492, Winkler).
Transmission electronic microscopy
C2C12 myotubes were dissociated with trypsin (Thermo Fisher Scientific), centrifuged, and fixed in 2% glutaraldehyde/0.1M cacodylate pH7.0. The cellular pellets were processed in the Advanced Microscopy Facility, Faculty of Biological Sciences at Pontificia Universidad Católica de Chile. Grids were visualized in Tecnai T12 microscopy (Philips). Mitochondrial morphology was analyzed by the ImageJ software
Data analysis
All results are expressed as mean and standard deviation. Statistical analyses were performed using two-way ANOVA to evaluate the effects of the LPS and UA factors, as well as their interaction. In case a main or interaction effect was found, a Sidak test was used as a post-hoc test. All statistical analyses were performed with GraphPad prism 9.3.0 analysis software. The p-values < 0.05 were considered statistically significant.
Results
Urolithin A reduces LPS-induced activation of the TLR4 pathway
TLR4 protein expression was unaffected by UA (p=0.69), LPS (p=0.53) or the interaction between LPS and UA (p=0.25) (Fig. 1A). MyD88 protein expression was unaffected by UA (p=0.36) and LPS (p=0.15) alone but tended to interact (p=0.07) (Fig. 1B). Downstream of TLR4, p-TAK was not regulated by UA (p=0.12), while a trend to upregulation was observed for LPS (p=0.06) (Fig. 1C). An interaction between UA and LPS was observed as well (p=0.03). Post-hoc analysis revealed that p-TAK was lower in UA+LPS compared to LPS alone (p=0.0351). p-IKKαβ tended to be up-regulated by LPS (p=0.07) and down-regulated by UA (p=0.062), without any interaction between both factors (p=0.74) (Fig. 1D). Similarly to p-TAK, IκBα protein expression was not modified by UA (p=0.19), while an upregulation by LPS was observed (p=0.050) (Fig. 1E). No interaction between LPS and UA was present (p=0.24). p65 NF-κB protein expression was not modified by UA (p=0.51) or LPS (p=0.76) alone nor in combination (p=0.32) (Fig. 1F). p-ERK was higher after LPS (p=0.003) but lower after UA incubation (p=0.02), with a significant interaction between LPS and UA (p=0.03) (Fig. 1G). Post-hoc analysis showed that p-ERK was lower after UA+LPS compared to LPS alone (p=0.0215). p-p38 was higher after LPS (p=0.04) but was not modify by UA alone (p=0.11) (Fig. 1H). Post-hoc analysis revealed that p-p38 was higher after LPS compared to CTL (p=0.05). When LPS and UA were combined, a tendency to an interaction effect was found (p=0.06). Finally, p-JNK was higher after LPS (p=0.006) but lower after UA incubation (p=0.01), with a significant interaction between both factors (p= 0.03) (Fig. 1I). Post-hoc analysis showed that p-JNK was lower when UA and LPS were combined compared to LPS alone (p=0.0319).
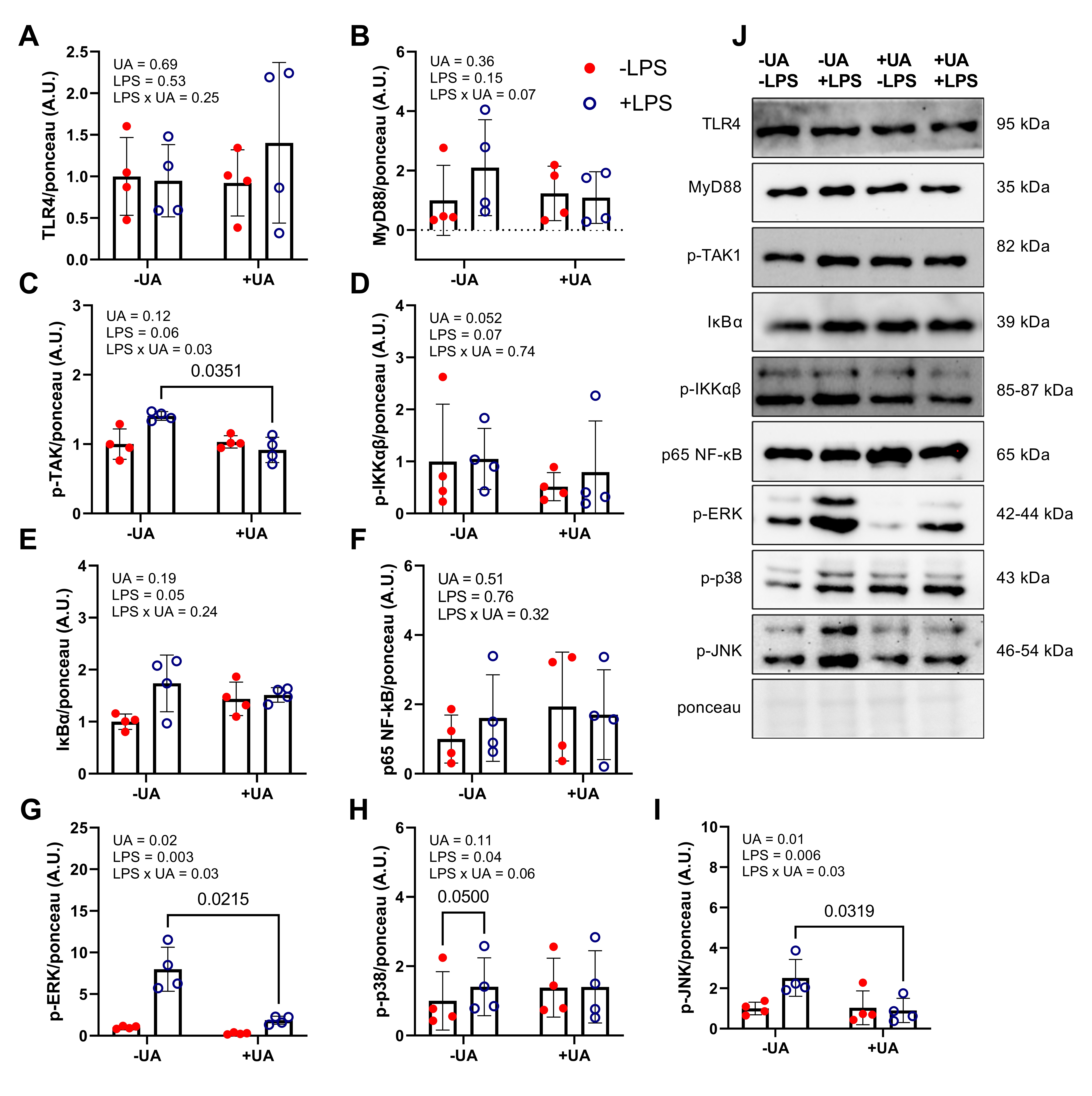
Fig. 1: Regulation of the TLR4 pathway. A-I) Protein expression and phosphorylation status of main components of the TLR4 pathway after incubation with urolithin A (UA) and lipopolysaccharide (LPS) alone or in combination in C2C12 myotubes. J) Representative blots. All data are reported as mean and standard deviation. Main (UA and LPS) and interaction (UA x LPS) effects are reported at the top left of each graph. Statistical difference reported by the post-hoc test is given above the concerned conditions.
Urolithin A prevents some alterations in mitochondrial morphology induced by LPS
Cellular components associated with lysosomal processes were observed by electron microscopy after LPS and after UA incubation alone but not in the CTL conditions and when UA and LPS were combined (Fig. 2A). LPS incubation decreased mitochondrial area (p=0.05) (Fig. 2B) and tended to decrease the perimeter (p=0.07) (Fig. 2C). For the perimeter, an interaction between UA and LPS was observed (p=0.02). Post-hoc analysis revealed that mitochondrial perimeter was lower in LPS compared to CTL (p=0.0314). UA (p=0.04) and LPS (p=0.0004) decreased mitochondrial circularity, with an interaction between both factors (p=0.01) (Fig. 2D). Finally, the mitochondrial aspect ratio was higher after UA (p=0.005) but was not modified by LPS alone (p=0.15) or when UA and LPS were combined (p=0.14) (Figure. 2E).
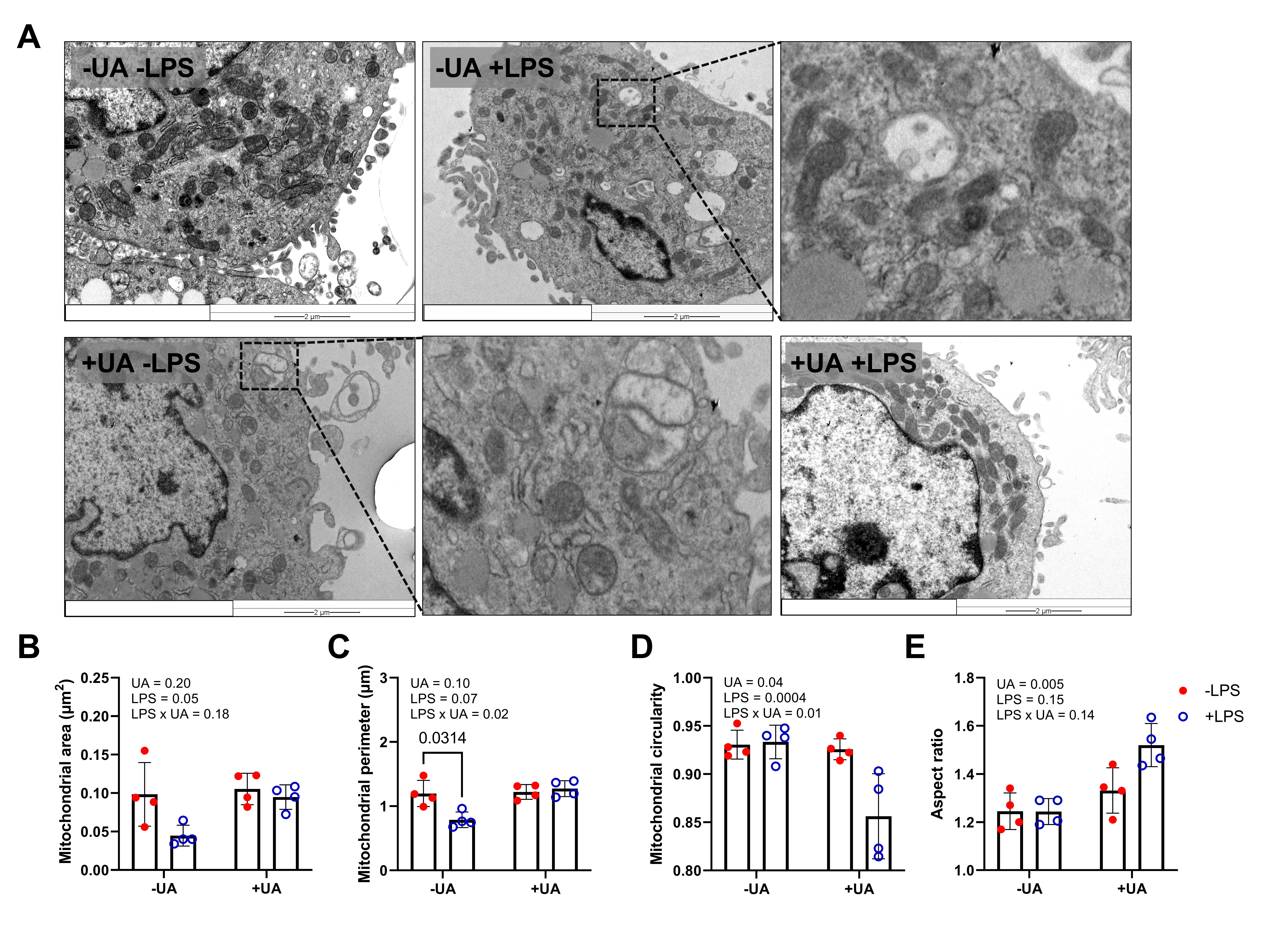
Fig. 2: Mitochondrial morphology. A) Representative images of mitochondrial morphology after incubation with urolithin A (UA) and lipopolysaccharide (LPS) alone or in combination in C2C12 myotubes using electron microscopy. B-E) Quantitative analysis of mitochondrial morphology. All data are reported as mean and standard deviation. Main (UA and LPS) and interaction (UA x LPS) effects are reported at the top left of each graph. Statistical difference reported by the post-hoc test is given above the concerned conditions.
UA decreases mitochondrial fission induced by LPS
The mitochondrial fission marker p-DRP1 was higher with LPS (p=0.009) but lower with UA (p=0.04), with no interaction between UA and LPS (p=0.46) (Fig. 3A). Post-hoc analysis indicated that p-DRP1 was lower when UA and LPS were combined compared to LPS alone (p=0.0363). The mitochondrial fusion marker Opa1 was not modified by any condition (Fig. 3B).
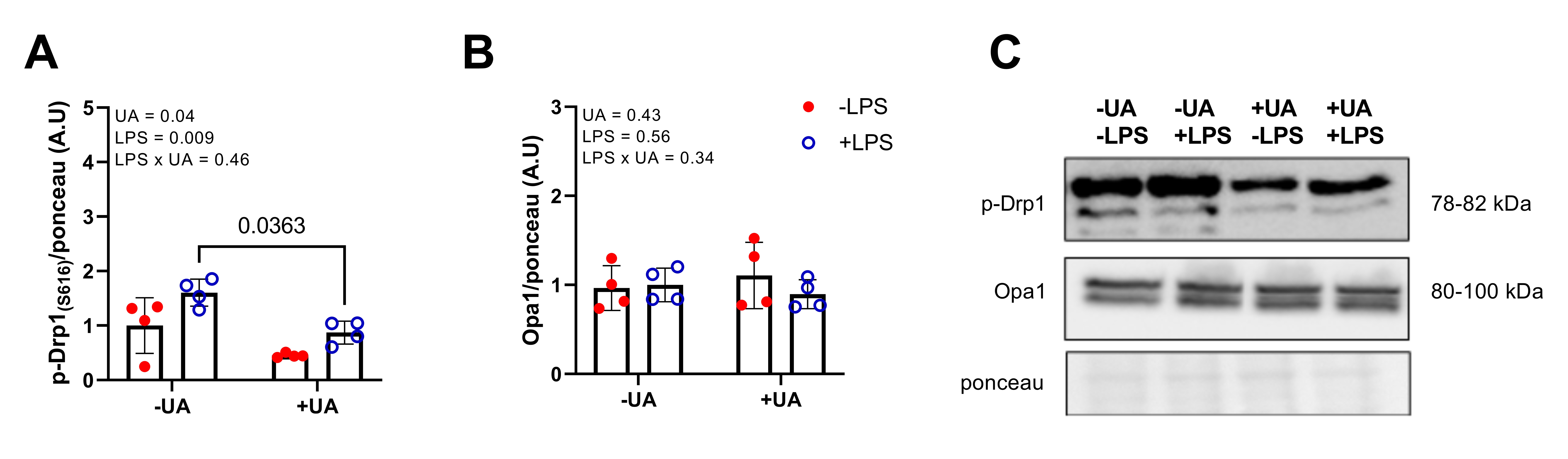
Fig. 3: Markers for mitochondrial dynamics. A) p-Drp1 and B) Opa1 protein expression after incubation with urolithin A (UA) and lipopolysaccharide (LPS) alone or in combination in C2C12 myotubes. C) Representative blots. All data are reported as mean and standard deviation. Main (UA and LPS) and interaction (UA x LPS) effects are reported at the top left of each graph. Statistical difference reported by the post-hoc test is given above the concerned conditions.
UA stimulates autophagy initiation
PINK1 protein expression was unaffected by UA (p=0.53) and LPS (p=0.73) alone, but there was an interaction between both factors (p=0.04) (Fig. 4A). Post-hoc analysis indicated a trend towards an upregulation of PINK1 protein expression between CTL and UA alone (p=0.08). A similar response was observed for p-AMPKα as no effect of UA (p=0.18) and LPS (p=0.43) alone was observed, while an interaction between both factors was present (p=0.03) (Fig. 4B). Post-hoc analysis revealed that p-AMPKα was higher in UA compared to CTL (p=0.03). Analysis of autophagy-related proteins revealed no differences in p62 levels (Fig. 4C) nor in ULK phosphorylation (Fig. 4D) in any condition. The LC3II/LC3I-II ratio was increased by UA (p=0.01) but remained unchanged after LPS incubation (p=0.27) (Fig. 4E). A tendency towards significance was observed for the interaction between LPS and UA (p=0.08). Post-hoc analysis revealed that the ratio was higher in UA than in CTL conditions (p=0.03).
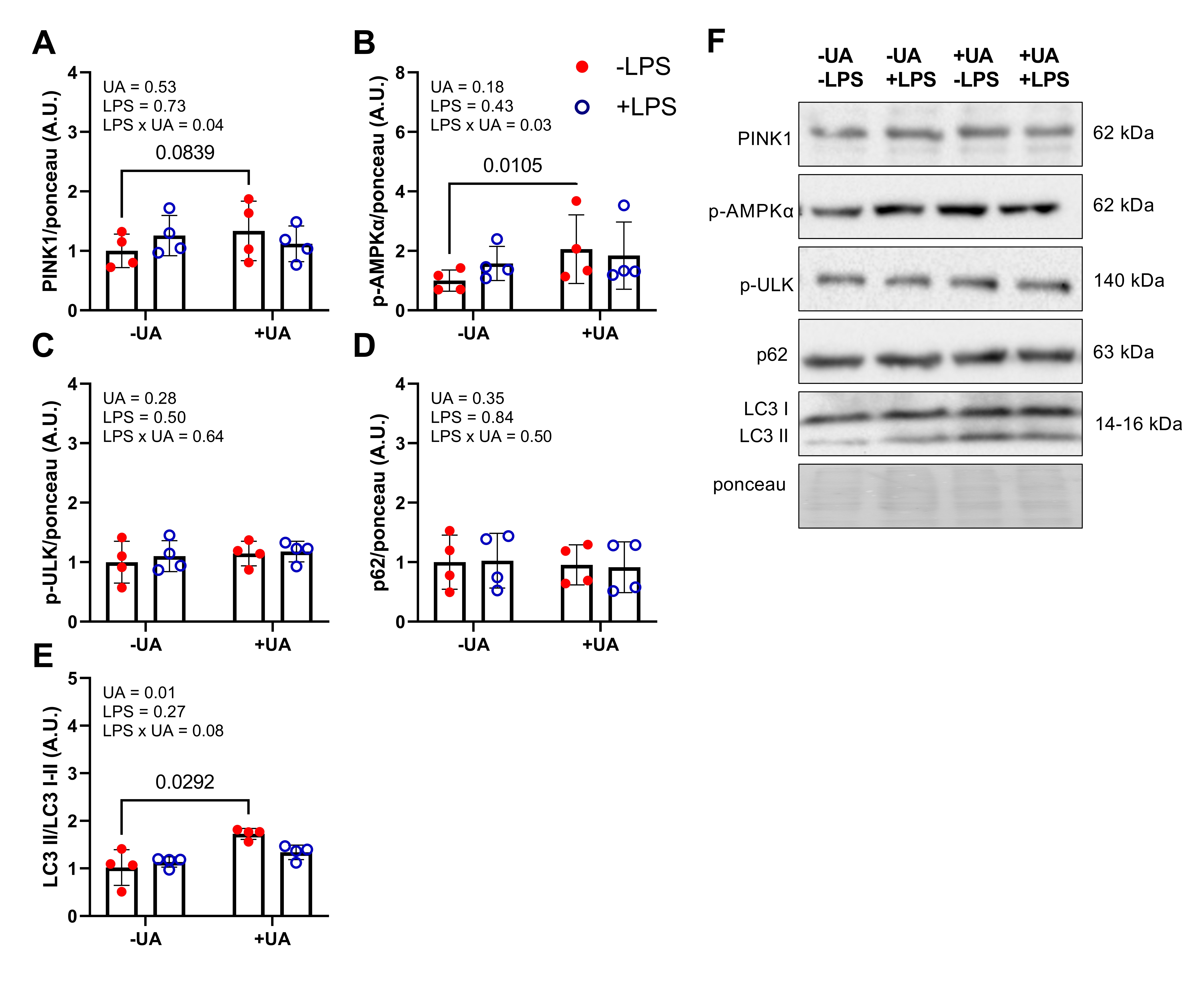
Fig. 4: Markers for the autophagy pathway. A-E) Protein expression and phosphorylation status of main components of the autophagy pathway after incubation with urolithin A (UA) and lipopolysaccharide (LPS) alone or in combination in C2C12 myotubes. F) Representative blots. All data are reported as mean and standard deviation. Main (UA and LPS) and interaction (UA x LPS) effects are reported at the top left of each graph. Statistical difference reported by the post-hoc test is given above the concerned conditions.
Discussion
The aim of the present study was to examine the effects of UA on key components and regulators of TLR4 signaling, mitochondrial dynamics and mitophagy, and to analyze mitochondrial morphology after LPS incubation in C2C12 myotubes. Here we found that: 1) LPS-induced inflammation activated downstream TLR4 signaling, by increasing the expression of IκBα and the phosphorylation of ERK, p38 and JNK and tended to increase the phosphorylation of TAK1 and IKKαb; 2) LPS reduced mitochondrial area and circularity and tended to reduce mitochondrial perimeter; 3) LPS increased the phosphorylation of the fission marker DRP1; 4) UA completely prevented, or at least attenuated, all previous effects induced by LPS. We first searched to confirm that LPS indeed activated the TLR4 pathway. We found that C2C12 incubation with 1µg LPS/ml for 3h increased the expression of IκBα and the phosphorylation of ERK, p38 and JNK and tended to increase the phosphorylation of TAK1 and IKKαb but no effect on p65 NF-κB expression was found. This partial activation of the TLR4 pathway is likely due to the dose of LPS used here. It was previously found that 1µg LPS/ml for 1h induced an increase in the DNA binding activity of NF-kB in C2C12 cells (16). Our mild activation of the TLR4 pathway by LPS could be prevented by pre-incubating cells for 21h with 50µM UA before adding LPS for an additional 3h. We chose the dose and duration of incubation based on the study of Ryu et al. reporting an increased mitophagy induced by UA in C2C12 cells (15). Our results are consistent with previous findings in chondrocytes. Specifically, 7.5 to 15µM UA was able to decrease the phosphorylation state of p38, ERK and JNK MAPK proteins following IL-1b-induced inflammatory response (14). This could indicate that the anti-inflammatory effects of UA are mediated, at least partially, by MAPK proteins. Beyond its role in inflammation, ERK has been found to play a key role in skeletal muscle by promoting a transition from fast to slow fibre type, which may have a protective effect in skeletal muscle diseases such as muscular dystrophy (17). Altogether, those results highlight the role of the MAPK family in pathological states. Knowing that the TLR4 signalling pathway regulates mitochondrial dynamics, specifically through MyD88-dependent activation and downstream activation of ERK (18, 19), we were interested in further investigating fusion and fission. We found that the phosphorylation of Drp1, which promotes mitochondrial fission or fragmentation (17, 20), was globally increased by LPS, but less so in the presence of UA. Thus, UA likely somewhat prevents LPS-induced Drp1 phosphorylation and thereby possibly mitochondrial fission. The exact mechanisms by which UA might regulate Drp1 cannot be evidenced based on our results. However, knowing that ERK phosphorylation displays a similar regulation pattern by LPS and UA and that ERK has been previously shown to regulate Drp1 (18), it can be speculated that this MAPK might contribute to the higher phosphorylation state of Drp1 observed in the present study. Of note, we did not find any change in Opa protein expression, a marker of protein fusion (21). Microscopy analyses revealed that the mitochondrial perimeter was reduced under LPS alone but not when pre-incubated with UA. Pre-treatment with UA thus prevented both the morphological alterations seen in the area and perimeter of mitochondria as well as the increase in phosphorylation of Drp1 following LPS stimulation. Any relation between the higher phosphorylation of Drp1 induced by UA and the protective effect observed on morphological alterations needs to be further investigated. Finally, as UA has been shown to induce some health, and more specifically muscle, benefits via an increase in mito- and autophagy (2, 22, 23), we investigated markers for autophagy under LPS stimulation in C2C12 cells. Additionally, it has been found that inflammation triggered by LPS might induce autophagy as a regulatory mechanism to control the inflammatory response (24). We were thus particularly interested in combining both UA and LPS and to investigate the autophagy pathway. However, our results showed that LPS-induced inflammation did not lead to changes in autophagy-related protein expression. As mentioned earlier, this could be due to the dose of LPS used in the present study and the absence of a frank activation of the TLR4/NF-κB pathway. Though, we found that UA increased the ratio of LC3II/LC3I-II, which might be indicative of a higher formation of autophagosomes. As the latter was not accompanied by any change in p62 expression, autophagic flux was probably not modified. Additionally, we found an interaction effect between UA and LPS for PINK1 protein expression and AMPKα phosphorylation. Our results are consistent with those reported by Ryu et al. (15), showing that UA induced mitophagy in C2C12 cells, primarily by increasing PINK1 protein expression and the LC3II/LC3I-II ratio, concomitantly to an increase in the phosphorylation of AMPKα. However, in the present study, we did not perform mitochondrial isolation, which limits our ability to directly associate the observed increase in autophagy-related proteins with mitophagy. In summary, the relation between UA, autophagy, and inflammation is still highly speculative but altogether, it could contribute to a better preservation of mitochondrial morphology under LPS stimulation. Although any causal relation needs to be further investigated. One limitation of in vitro studies is the translation of the findings to in vivo models and more specifically to human. Here, we used a dose of 50µM of urolithin A based on previous findings in C2C12 cells (15). In the same study, doses of 25 and 50mg/kg/d for several weeks to several months have been found to exert beneficial effects on muscle function in rodents (15). Shorter durations have been used as well, i.e. 3 and 15 days with a dose of 20mg/kg/d for 5 days per week, to reduce the deleterious effects of alcoholic chronic pancreatitis in mice (25). In human, improved health and physical outcomes have been observed with supplementation protocols of 4 months of 500mg of urolithin A in middle-aged adults (22) and even more so with 1000mg in middle-aged (22) and older people (23, 26). While in vitro dosage and duration are difficult to translate to in vivo protocols, a dose of a few hundred milligrams of urolithin A has been consistently used in human and is considered as safe (27).
Conclusion
Our findings suggest that UA partially mitigates LPS-induced inflammation by modulating the TLR4 signaling pathway and mitochondrial dynamics. Even though no causal relation may be established based on the present findings, UA may represent a promising therapeutic agent for conditions associated with mitochondrial dysfunction and inflammation, highlighting its potential role in promoting skeletal muscle health.
Acknowledgements
Author Contributions
HZF and GTC designed the study; GTC and MVB acquired the data; GTC, MVB, LD and HZF analyzed and interpreted the results; GTC, LD and HZF wrote the first draft; all authors approved the final version of the manuscript.
Funding Sources
This study was funded by a chair of investigation on muscular diseases of the Universidad Finis Terrae.
Statement Of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare. Artificial intelligence has not been used at any stage of the present work.
References
- Tomás-Barberán FA, González-Sarrías A, García-Villalba R, Núñez-Sánchez MA, Selma MV, García-Conesa MT, et al. Urolithins, the rescue of "old" metabolites to understand a "new" concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol Nutr Food Res. 2017;61(1). https://doi.org/10.1002/mnfr.201500901
- D'Amico D, Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol Med. 2021;27(7):687-99. https://doi.org/10.1016/j.molmed.2021.04.009
- Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013-22. https://doi.org/10.1038/s41556-018-0176-2
- Faitg J, D'Amico D, Rinsch C, Singh A. Mitophagy Activation by Urolithin A to Target Muscle Aging. Calcif Tissue Int. 2024;114(1):53-9. https://doi.org/10.1007/s00223-023-01145-5
- Luan P, D'Amico D, Andreux PA, Laurila PP, Wohlwend M, Li H, et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci Transl Med. 2021;13(588). https://doi.org/10.1126/scitranslmed.abb0319
- Picca A, Faitg J, Auwerx J, Ferrucci L, D'Amico D. Mitophagy in human health, ageing and disease. Nat Metab. 2023;5(12):2047-61. https://doi.org/10.1038/s42255-023-00930-8
- Tapia-Curimil G, Castro-Sepulveda M, Zbinden-Foncea H. Effect of epicatechin consumption on the inflammatory pathway and mitochondria morphology in PBMC from a R350P desminopathy patient: A case report. Physiol Rep. 2024;12(8):e16020. https://doi.org/10.14814/phy2.16020
- Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther. 2023;8(1):333. https://doi.org/10.1038/s41392-023-01547-9
- Han H, Tan J, Wang R, Wan H, He Y, Yan X, et al. PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020;21(8):e48686. https://doi.org/10.15252/embr.201948686
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221-5. https://doi.org/10.1038/nature09663
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145-51. https://doi.org/10.1016/j.cyto.2008.01.006
- Castro-Sepulveda M, Tuñón-Suárez M, Rosales-Soto G, Vargas-Foitzick R, Deldicque L, Zbinden-Foncea H. Regulation of mitochondrial morphology and cristae architecture by the TLR4 pathway in human skeletal muscle. Front Cell Dev Biol. 2023;11:1212779. https://doi.org/10.3389/fcell.2023.1212779
- Tao H, Li W, Zhang W, Yang C, Zhang C, Liang X, et al. Urolithin A suppresses RANKL-induced osteoclastogenesis and postmenopausal osteoporosis by, suppresses inflammation and downstream NF-κB activated pyroptosis pathways. Pharmacol Res. 2021;174:105967. https://doi.org/10.1016/j.phrs.2021.105967
- Ding SL, Pang ZY, Chen XM, Li Z, Liu XX, Zhai QL, et al. Urolithin a attenuates IL-1β-induced inflammatory responses and cartilage degradation via inhibiting the MAPK/NF-κB signaling pathways in rat articular chondrocytes. J Inflamm (Lond). 2020;17:13. https://doi.org/10.1186/s12950-020-00242-8
- Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22(8):879-88. https://doi.org/10.1038/nm.4132
- Ono Y, Maejima Y, Saito M, Sakamoto K, Horita S, Shimomura K, et al. TAK-242, a specific inhibitor of Toll-like receptor 4 signalling, prevents endotoxemia-induced skeletal muscle wasting in mice. Sci Rep. 2020;10(1):694. https://doi.org/10.1038/s41598-020-57714-3
- Boyer JG, Prasad V, Song T, Lee D, Fu X, Grimes KM, et al. ERK1/2 signaling induces skeletal muscle slow fiber-type switching and reduces muscular dystrophy disease severity. JCI Insight. 2019;5(10). https://doi.org/10.1172/jci.insight.127356
- Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284(24):4177-95. https://doi.org/10.1111/febs.14122
- Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42(3):406-17. https://doi.org/10.1016/j.immuni.2015.02.002
- Kapetanovic R, Afroz SF, Ramnath D, Lawrence GM, Okada T, Curson JE, et al. Lipopolysaccharide promotes Drp1-dependent mitochondrial fission and associated inflammatory responses in macrophages. Immunol Cell Biol. 2020;98(7):528-39. https://doi.org/10.1111/imcb.12363
- Quintana-Cabrera R, Scorrano L. Determinants and outcomes of mitochondrial dynamics. Mol Cell. 2023;83(6):857-76. https://doi.org/10.1016/j.molcel.2023.02.012
- Singh A, D'Amico D, Andreux PA, Fouassier AM, Blanco-Bose W, Evans M, et al. Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell Rep Med. 2022;3(5):100633. https://doi.org/10.1016/j.xcrm.2022.100633
- Liu S, D'Amico D, Shankland E, Bhayana S, Garcia JM, Aebischer P, et al. Effect of Urolithin A Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults: A Randomized Clinical Trial. JAMA Netw Open. 2022;5(1):e2144279. https://doi.org/10.1001/jamanetworkopen.2021.44279
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255-63. https://doi.org/10.1038/ni.2215
- Mehra S, Srinivasan S, Singh S, Zhou Z, Garrido V, Silva IC, et al. Urolithin A attenuates severity of chronic pancreatitis associated with continued alcohol intake by inhibiting PI3K/AKT/mTOR signaling. Am J Physiol Gastrointest Liver Physiol. 2022;323(4):G375-G86. https://doi.org/10.1152/ajpgi.00159.2022
- Denk D, Singh A, Kasler HG, D'Amico D, Rey J, Alcober-Boquet L, et al. Effect of the mitophagy inducer urolithin A on age-related immune decline: a randomized, placebo-controlled trial. Nat Aging. 2025;5(11):2309-22. https://doi.org/10.1038/s43587-025-00996-x
- Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1(6):595-603. https://doi.org/10.1038/s42255-019-0073-4