BRCA2 Polymorphisms and Breast Cancer Susceptibility: a Multi-Tools Bioinformatics Approach
bDepartment of Allied Health Sciences, Faculty of Nursing, Al-Balqa Applied University (BAU), Al-Salt 19117, Jordan,
cDepartment of Biochemistry, Jamia Hamdard, Delhi, India,
dZoology Department, College of Science, King Saud University, P.O. Box: 2455, 11451, Riyadh, Saudi Arabia,
eCentre for Applied Mathematics and Bioinformatics (CAMB), Gulf University for Science and Technology, Hawally, Kuwait.
Keywords
Abstract
Background/Aims:
The main focus of this investigation is to identify deleterious single nucleotide polymorphisms (SNPs) located in the BRCA2 gene through in silico approach, thereby, providing an understanding of potential consequences regarding the susceptibility to breast cancer.Methods:
The GenomAD database was used to identify SNPs. To determine the potential adverse consequences, our study employed various prediction tools, including SIFT, PolyPhen, PredictSNP, SNAP2, PhD-SNP, and ClinVar. The pathogenicity associated with the deleterious snSNPs was evaluated bu MutPred and Fathmm. Additionally, I-Mutant and MuPro were used to assess the stability, followed by conservation and protein-protein interaction analysis using robust computational tools. The 3D structure of BRCA2 protein was generated by SwissModel, followed by validation using PROCHECK and Errat.Results:
The GenomAD database was used to identify a total of 7, 921 SNPs, including 1940 missense SNPs. A set of 69 SNPs predicted by consensus to be damaging across all platforms was identified. Mutpred and Fathmm identified 48 and 38 SNPs, respectively to be associated with cancer. While I- Mutant and MuPro assays suggested 22 SNPs to decrease protein stability. Additionally, these 22 SNPs reside within highly conserved regions of the BRCA2 protein. Domain analysis, utilizing InterPro, pinpointed 18 deleterious mutations within crucial DNA binding domains and one in the BRC repeat region.Conclusion:
This study establishes a foundation for future experimental validations and the creation of breast cancer-targeted treatment approaches.Introduction
The BRCA2 gene, also known as Breast Cancer 2 susceptibility protein, plays a crucial role in maintaining the integrity of the cell’s DNA. It is involved in the repair of damaged DNA and has a key function in suppressing tumor formation. Mutations in the BRCA2 gene are associated with an increased risk of developing breast and ovarian cancers [1]. These mutations are inherited in an autosomal dominant pattern, meaning that individuals with a mutation in one copy of the BRCA2 gene have a significantly higher probability of developing these types of cancer [2].
It has been estimated that approximately 50–60% of women with a germline BRCA2 mutation will develop breast cancer during their lifetime [3]. Furthermore, individuals with a BRCA2 mutation also have a 30% increased risk of developing ovarian cancer [4]. In addition to breast and ovarian cancer, the BRCA2 gene mutation has been linked to an increased risk of other types of malignancies. For example, men with a BRCA2 mutation have an increased risk of developing breast cancer and prostate cancer [5] which further emphasizes the importance of recognizing and understanding the association between the BRCA2 gene and cancer development. Variations in the BRCA2 gene can have clinical implications for both the diagnosis and treatment of breast cancer [6].
The BRCA2 gene is located on chromosome 13 and consists of 27 exons [7] each of these exons plays a crucial role in encoding the BRCA2 protein, which is comprised of 3, 418 amino acids [8] with the Uniprot ID: P51587 [9]. BRCA2 includes three domains. These domains are involved in binding to single-stranded DNA (ssDNA). These domains are: Oligonucleotide/Oligosaccharide-Binding (OB) Domains- BRCA2 contains multiple OB domains, which are involved in recognizing and binding to single-stranded DNA (ssDNA) [10]; BRC Tandem Repeats- play a role in promoting protein-protein interactions [11]; TR2 C-Terminal Domain- The TR2 C-terminal domain refers to a specific region at the C-terminus of the BRCA2 protein, which possibly contributes to interactions with other cellular components [12][13]. The DNA binding domain of the BRCA2 protein is of particular interest in understanding its role in breast cancer. This domain is responsible for interacting with other proteins and DNA molecules, allowing for proper DNA repair and maintenance of genomic stability. One such protein that interacts with the DNA binding domain of BRCA2 is DSS1. DSS1, also known as deleted in split hand/split foot 1, is a small protein that has been found to interact with the DNA binding domain of BRCA2. This interaction is thought to be important for the stability and function of the BRCA2 protein in DNA repair processes [14]. The BRCA2 gene also contains eight conserved motifs called BRC repeats that are involved in binding to the RAD51 protein [15].
In recent years, researchers have been using bioinformatics tools to predict the mutation which might be harmful without the need for time consuming and expensive lab experiments. These predicted harmful changes are then considered for further experimental analysis. Our study takes this approach by using a variety of computer-based tools, each based on different principles, to examine whether certain changes in the BRCA2 might be harmful. We utilized bioinformatics tools to identify a comprehensive list of SNPs within the BRCA2 gene and apply computational analysis to assess the potential pathogenicity of the identified SNPs, considering factors such as conservation, functional impact, and protein structure disruption. Instead of confirming these changes through experiments, we aim to offer a quick and cost-effective method for identifying potentially problematic genetic variations.
Materials and Methods
The comprehensive methodology, encompassing the tools and databases utilized to discern deleterious SNPs within the human BRCA2 gene, is concisely outlined in Fig. 1.
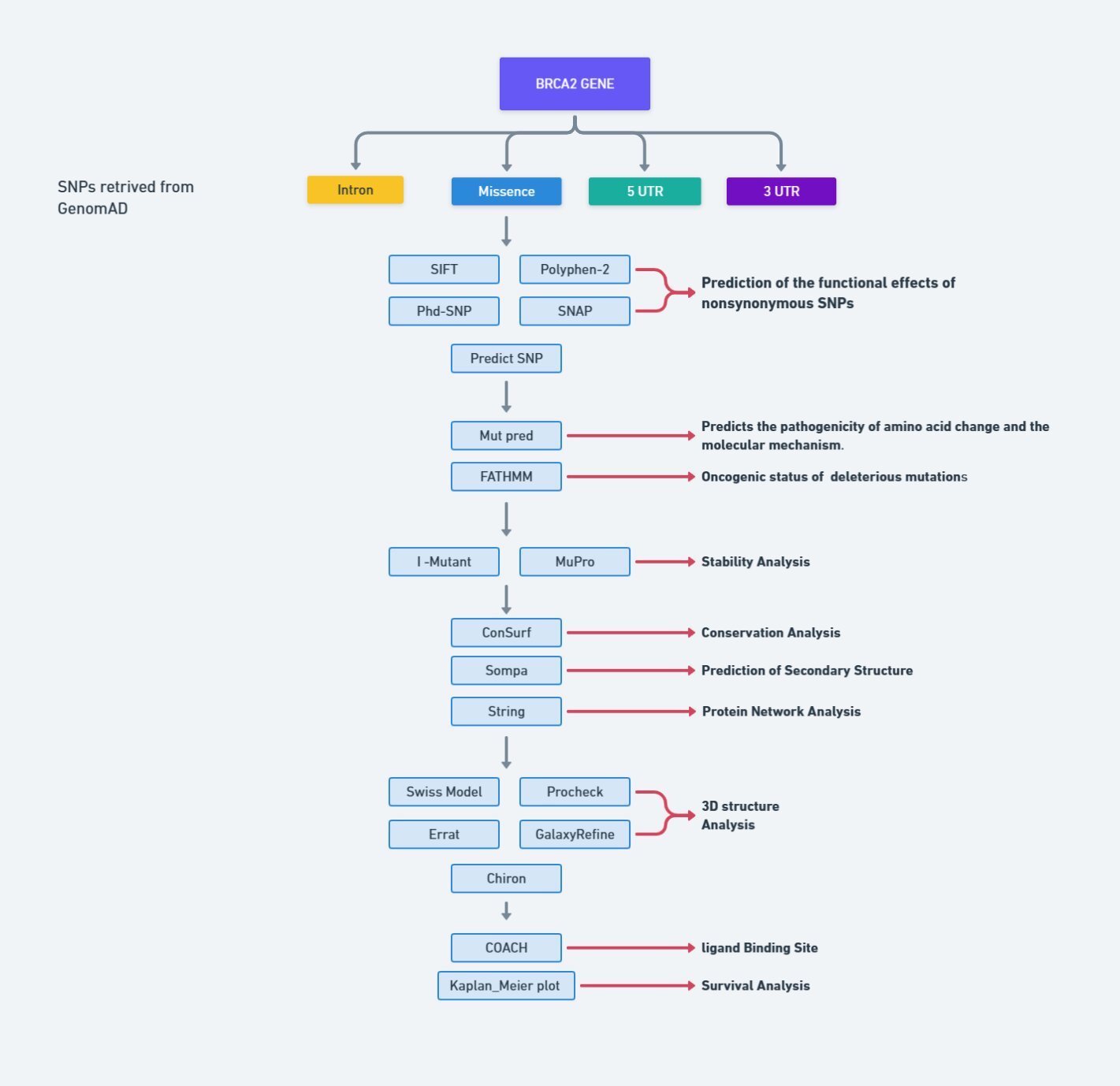
Fig. 1: Flowchart for methodology.
Retrieval of BRCA2 nsSNPs
We utilized the Genome Aggregation Database () for the retrieval of single nucleotide polymorphisms for the BRCA2 gene. Only missense SNPs within the BRCA2 gene were selected because they have the potential to impact the structural and functional characteristics of the BRCA2 protein. The FASTA sequence of BRCA2 protein was acquired from the Uniprot web server.
Identifying the most damaging nsSNPs
To identify the most damaging non-synonymous single nucleotide polymorphisms (nsSNPs) within the BRCA2 gene, we employed a comprehensive approach using five distinct bioinformatics tools. These computational programs encompassed: SIFT (Sorting Intolerant from Tolerant) [https://sift.bii.a-star.edu.sg/], PolyPhen-2 (Polymorphism Phenotyping v2) [http://genetics.bwh.harvard.edu/pph2/], PhD-SNP (Predictor of human Deleterious Single Nucleotide Polymorphisms) [http://snps.biofold.org/phd-snp/phd-snp.html] predict SP, SNAP [16–19]. Additionally, the pathogenic mutations were further evaluated using ClinVar (). ClinVar, is a comprehensive database that aggregates variant interpretations from various sources, including clinical laboratories and expert panels, categorizing variants as “benign,” “likely benign,” “uncertain significance,” “likely pathogenic,” and “pathogenic” based on available evidence [20]. SIFT, on the other hand, assesses the evolutionary conservation of amino acids across species, predicting whether an amino acid substitution is tolerated or likely to be deleterious based on conservation trends [21]. PolyPhen-2 classifies variants as “probably damaging,” “possibly damaging,” or “benign” by integrating sequence- and structure-based features and taking into account factors such as sequence conservation, physicochemical properties, and structural information [22]. PhD-SNP uses support vector machines to distinguish between disease-associated and neutral nsSNPs by analyzing sequence-derived features such as conservation, physicochemical properties, and structural characteristics [23]. Subsequently, we categorized nsSNPs as high risk if they were predicted as deleterious, Variant of Uncertain Significance (VUS) and pathogenic by all these computational tools. These high risk nsSNPs were then selected for further analysis.
MutPred: Predicting Disease Associated Amino Acid Substitutions and Phenotypic Outcomes
We used the MutPred web server (http://mutpred.mutdb.org/) as a valuable tool for understanding the molecular mechanisms underlying diseases triggered by amino acid substitutions within mutant proteins. MutPred employs an array of structural, functional, and evolutionary protein features to deliver insights into the consequences of these substitutions [24]. MutPred2 predicts the pathogenicity of amino acid substitutions and their molecular mechanisms, considering factors like changes in protein stability, potential loss of catalytic sites, and the possibility of gaining O-linked glycosylation sites. To achieve this, MutPred integrates the power of various tools and algorithms, including PSI-BLAST, SIFT, Pfam profiles, TMHMM, MARCOIL, and DisProt. These computational methods collectively provide projections regarding structural damage caused by amino acid substitutions.
Prediction of Cancer Linked Mutations
As part of our research on the association between the BRCA2 gene and breast cancer, we sought to identify mutations that may be linked to cancer development. To accomplish this, we utilized the Functional Analysis through Hidden Markov Models (Fathmm) webserver () [25]. Fathmm is a valuable resource that integrates sequence conservation information within hidden Markov models (HMMs). The Fathmm server is widely recognized for its high throughput capabilities in predicting the phenotypic, molecular, and functional consequences of protein variants within both coding and noncoding regions [26]. It employs a combination of unweighted and weighted algorithms, incorporating sequence conservation alongside pathogenicity weights to make predictions. In the context of our study, it is important to note that the Fathmm server employs a default prediction threshold of -0.75. Mutations with a prediction score lower than this threshold are considered potentially associated with cancer. This step was particularly relevant for our research as it enabled us to identify SNPs within BRCA2 that may have implications for breast cancer development.
Prediction of protein stability change
Assessing the stability of a protein is a crucial aspect in understanding its functionality within biological processes. The stability of a protein is intricately tied to its folding energy, with mutations having the potential to directly impact this energy and consequently, stability. To predict the effect of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein stability, we employed two computational tools: I-MUTANT 3.0 server ( 2.0.html) and MUPRO (). These tools utilize support vector machine (SVM) models to make predictions. In our analysis, a thermodynamics free energy value greater than 0 was indicative of protein stability, while a negative value signified destabilization. This approach allowed us to determine how nsSNPs may influence the stability of the protein.
Conservation analysis
To estimate evolutionary conservation of specific amino acid positions within the protein of interest, we employed ConSurf, a bioinformatics tool designed for precisely this purpose [27]. ConSurf’s methodology relies on examining the evolutionary relationships among homologous sequences. We estimated the conservation levels of amino acid residues by considering a set of 50 homologous sequences. Notably, we identified amino acid positions that exhibited a high degree of conservation. These residues were chosen for further analysis.
Secondary Structure Prediction
To predict the amino acid secondary structure of BRCA2, we utilized the SOPMA tool, a widely recognized resource for analyzing protein secondary structure. While other tools like PSipred exist for this purpose, we opted for SOPMA due to its capability to handle long amino acid sequences such as BRCA2. SOPMA employs a robust algorithm to estimate the distribution of secondary structure elements, including alpha helices, beta sheets, and random coils [28, 29]. The results from this analysis provided valuable insights into the secondary structure composition of the BRCA2 protein.
Protein-Protein Interaction
In the realm of proteins, understanding their interactions is crucial to unravel their roles in disease processes. To investigate the intricate web of protein-protein interactions involving BRCA2, we harnessed the STRING tool (Search Tool for the Retrieval of Interacting Genes) [30]. This powerful resource unveiled the network of connections, shedding light on BRCA2’s collaborations with other molecules.
Identification of a deleterious mutation in the functional domain
The functional domain of the BRCA2 was identified using InterProScan, a useful tool accessible at [31] InterProScan integrates various methods for detecting unique protein signatures from the InterPro consortium’s associated databases into a single resource. This web-based version of InterPro is available for both academic and commercial organizations through European Bionformatics Institute (EBI). To perform this analysis, we utilized the InterProScan tool, which scans protein sequences provided in FASTA format to find matches in the InterPro protein signature databases.
Modeling of Protein Structure
In the process of modeling the protein structure of BRCA2, we obtained the FASTA sequence from the Uniprot Protein Database. To generate the 3D structure, we utilized the Swiss Model web server, which is renowned for producing reliable structural models [32]. This server offers valuable insights through QMEAN scores and the alignment with experimental protein sequences. To enhance the predicted structure’s quality, we applied refinement techniques via GalaxyRefine and conducted a comprehensive structural assessment using PROCHECK and Errat. Additionally, mutant models were manually crafted based on the predicted protein structure with the assistance of PyMol. These models were energy minimized via Swiss PDB viewer and then with Chiron. RMSD of mutated models was calculated via TM alignment tool.
Ligand Binding Site Prediction
To identify active sites and potential ligand-binding locations within BRCA2, we employed the COACH protein-ligand binding prediction server. This meta-server approach, available at http://zhanglab.ccmb.med.umich.edu/COACH/, offers a comprehensive analysis of protein-ligand binding sites. We utilized a model generated by Swiss-Model as input for COACH to predict these crucial ligand-binding regions. The top 10 models were ranked by the cluster size and assigned a C-score, which ranged from 0 to 1, where higher scores indicate increased reliability.
Correlation of BRCA2 Deregulation with Breast and Ovarian Cancer
The clinical significance of BRCA2 was assessed in relation to the survival of breast cancer patients, utilizing the Kaplan–Meier plotter available at https://kmplot.com/analysis/. This comprehensive tool has evaluated the expression of 54, 000 genes across 21 cancer types, drawing data from over 10, 000 patient samples, including 6, 234 individuals with breast cancer. The system incorporates diverse sources such as the Gene Expression Omnibus (GEO), European Genome-Phenome Archive (EGA), and The Cancer Genome Atlas (TCGA). Through this extensive analysis, we gained valuable insights into the correlation between BRCA2 expression and the survival outcomes of breast cancer patients. The prediction of the comprehensive survival rate of various types of cancer patients, i.e. breast, and ovarian was done with Kaplan–Meier plot. The survival analyses of cancer patients were run against, 4929, 1435, breast and ovarian patients, respectively.
Results
Retrieval of nsSNPs
A total of 7, 921 SNPs were retrieved from the GenomAD database, representing a diverse array of genetic variations. Within this dataset, there were distinct categories of SNPs, including 1, 940 missense or non-synonymous SNPs, 5, 887 intronic SNPs, 54 SNPs located in the 3-prime untranslated regions (UTR), and 40 SNPs present in the 5-prime UTR regions. In our subsequent analysis, we specifically focused on the non-synonymous SNPs for further investigation.
Identifying the most damaging nsSNPs
In our comprehensive SNP analysis, we employed five distinct prediction tools, namely SIFT, PolyPhen, PredictSNP, SNAP2, PhD-SNP, and ClinVar, to assess the potential deleterious impact of 1940 SNPs. The results revealed that a substantial number of SNPs were classified as deleterious by these tools, with SIFT identifying 622 SNPs, PolyPhen identifying 793 SNPs, PredictSNP identifying 518 SNPs, SNAP2 identifying 639 SNPs, and PhD-SNP identifying 151 SNPs as deleterious (Fig. 2). Importantly, these predictions served as a basis for identifying a consensus set of 69 SNPs that demonstrated deleterious effects across all five prediction tools (Table S1). Among the identified 69 SNPs, we used ClinVar and Varsome to assess their clinical significance. Of these, 16 were classified as pathogenic, conversely, 11 SNPs were categorized as benign. Interestingly, a substantial portion, comprising 42 SNPs, fell into the category of variants of uncertain significance (VUS). It’s noteworthy that despite this variability in ClinVar classification, the selection criteria for our further analysis remained stringent, focusing exclusively on the 69 SNPs unanimously deemed deleterious by all five prediction tools, ensuring a high level of confidence in their potential functional impact. These 69 SNPs, exhibiting unanimity in their deleterious predictions, were selected for further in-depth analysis in our study.
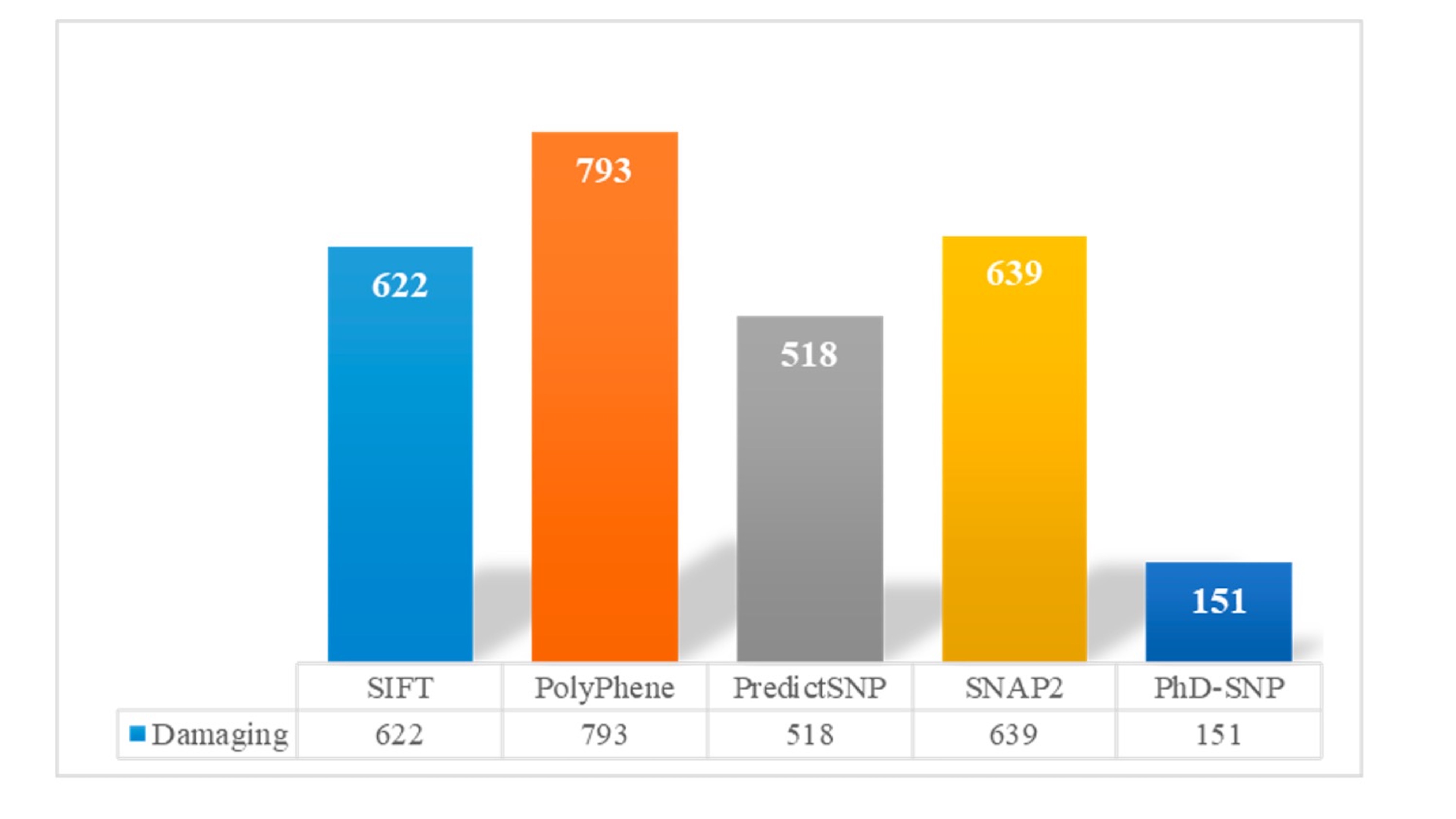
Fig. 2: Showing damaging nsSNPs identified by five In-silico tools; SIFT, Predict-SNP, PolyPhen-2, Phd-SNP and SNPs&GO.
Predicting Disease-Associated Amino Acid Substitutions and Phenotypic Outcomes
To predict disease-associated amino acid substitutions and phenotypic outcomes, we used MutPred, a valuable analytical tool. Among the 69 selected missense SNPs subjected to MutPred analysis, a noteworthy subset of 48 SNPs exhibited a substantial probability of damaging the associated proteins, as indicated by MutPred scores exceeding the critical threshold of 0.5 (Table S2). This careful curation based on scores led to the identification of 48 SNPs that were deemed to have a higher likelihood of influencing molecular mechanisms and contributing to disease pathogenesis. These SNPs, characterized by their potential to disrupt protein function, were subsequently chosen for an in-depth and focused investigation in our research, shedding light on their potential roles in disease etiology and offering valuable insights for further exploration.
Prediction of Cancer-Linked Mutations
In the pursuit of identifying mutations with potential links to cancer, our study applied a comprehensive Functional Analysis through Hidden Markov Models (Fathmm) using the carefully selected 48 SNPs. Employing a stringent threshold value of -0.75, we discerned that 38 of these SNPs exhibited Fathmm scores below this critical threshold (Table 1). This observation strongly suggests an association between these 38 SNPs and cancer, prompting their selection for further in-depth analysis. By leveraging Fathmm’s predictive capabilities, we have identified a subset of mutations that warrant closer examination due to their potential implications in cancer-related molecular mechanisms, offering a promising avenue for subsequent research endeavors in this vital area of study.
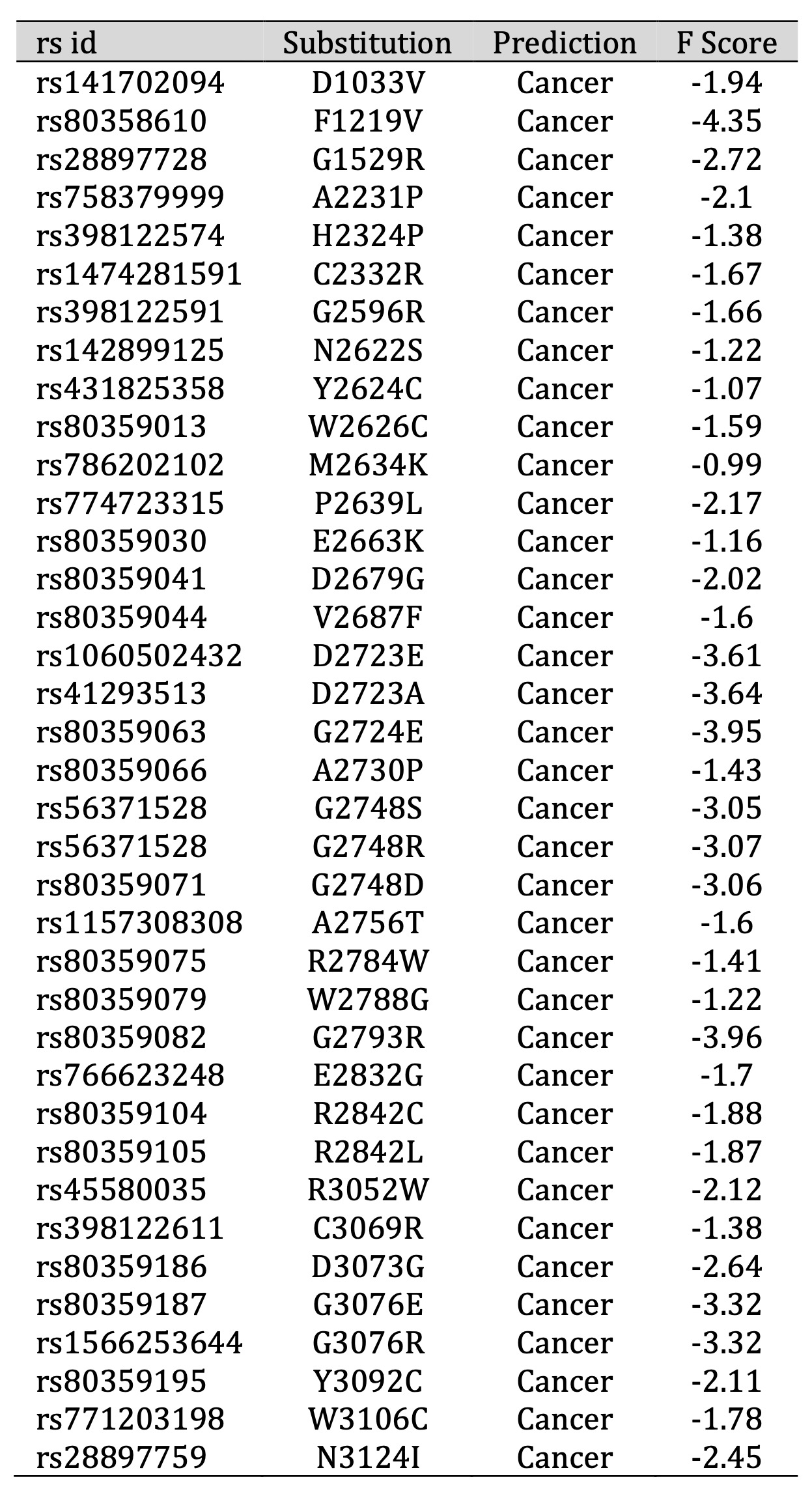
Table 1: Predicting cancer and disease-associated amino acid substitution using Fathmm
Prediction of protein stability change
To predict the stability of proteins influenced by specific genetic mutations, we focused on 38 particular SNPs (Single Nucleotide Polymorphisms) that have potential implications in cancer. Employing computational tools like I-Mutant and MuPro, we conducted a thorough analysis. The outcome of this analysis unveiled that 22 of the 38 exhibited a negative SVM (Support Vector Machine) score, negative ΔΔG (Free Energy Change) and negative NN (Neural Network) values indicating a decrease in protein stability (Table 2). This decline in stability can disrupt protein functions, making these mutations potentially more harmful. The protein stability of 22 mutations with respect to free energy values via I-Mutant showed that all 22 mutant models had free energy change (ΔΔG) values ranging between -3.911 and -0.22, predicting a decrease in stability in forming a protein structure.
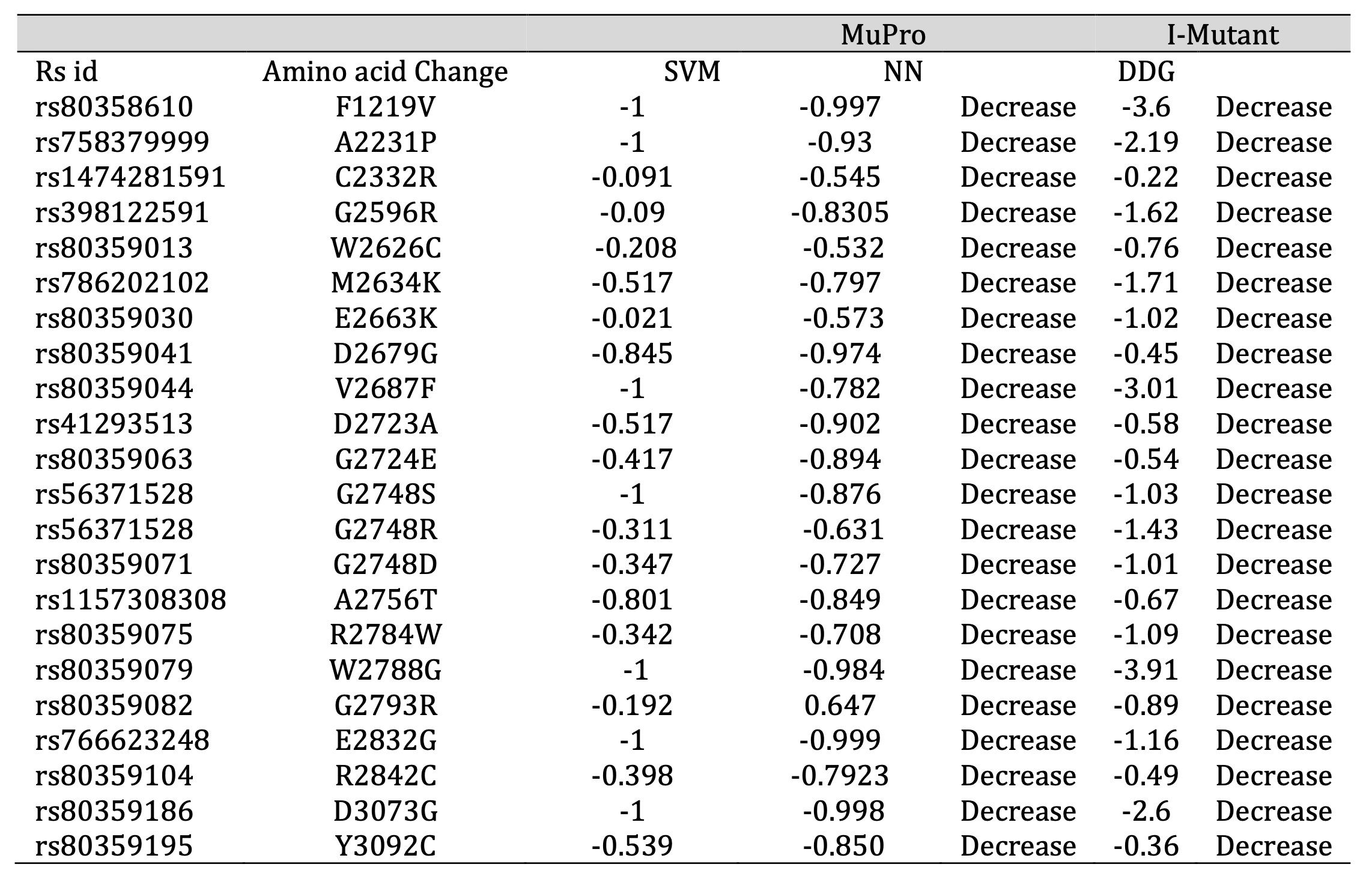
Table 2: Predicted Alterations in Protein Stability by I-MUTANT 2 and Mupro
Conservation analysis
We explored the evolutionary conservation aspect of our study by employing the ConSurf web browser to assess the level of conservation at each position within the BRCA2 protein. Through its Bayesian methodology, ConSurf can pinpoint potentially critical amino acids, shedding light on both their functional and structural significance based on their evolutionary conservation profiles. Intriguingly, after subjecting all 22 SNPs to ConSurf analysis, we found that they reside within highly conserved regions, boasting impressive scores ranging between 8 and 9 (Table 3). This discovery reinforces their importance and potential impact, prompting us to include them in our subsequent in-depth analyses. These SNPs, nestled within regions that have stood the test of evolutionary time, may harbor valuable clues regarding their roles in disease etiology and protein function.
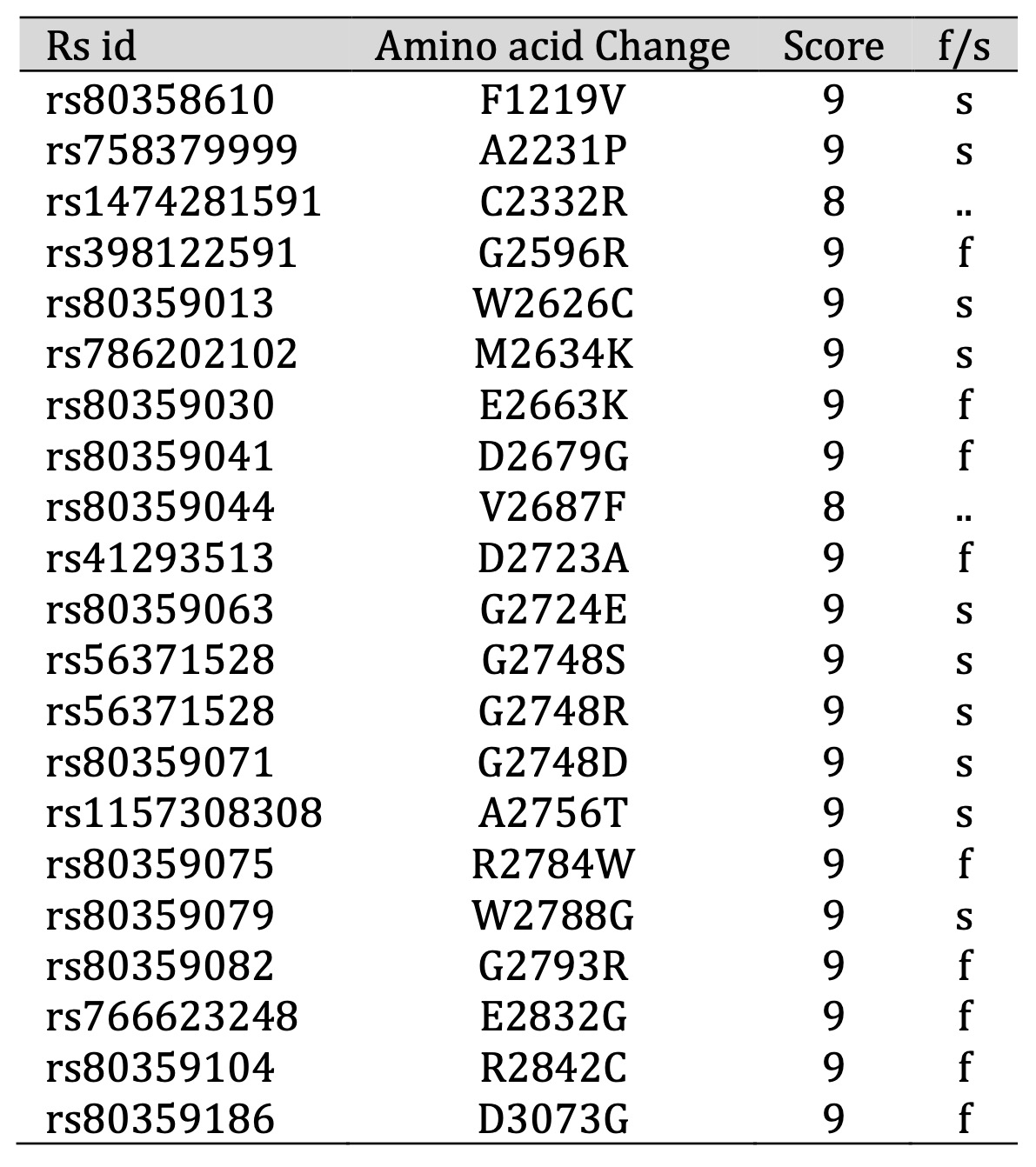
Table 3: Prediction of evolutionary conservation by ConSurf. According to the ConSurf server, “f” — functional residue and “s” — structural residue
Prediction of BRCA2 Secondary Structure
To forecast the secondary structure of BRCA2, we employed the SOPMA tool in our analysis elucidating the prevalence of alpha helices, beta sheets, and coil structures. The analysis unveiled a prominent presence of random coils, accounting for 52.52% (1795) of the secondary structure, followed by 28.94% (989) in alpha helices, 14.80% (506) in extended strands, and 3.74% (128) in beta turns within the predicted secondary structure of BRCA2.
Analyzing Protein Interactions
Protein interactions are vital for various cellular functions, including cell signaling. Given this intricate network, any variations in amino acids within a protein can potentially influence the behavior of other proteins in the network. Our exploration of BRCA2’s functional partners using the STRING tool revealed a group of ten significant proteins: PALB2, SEM1, BRIP1, RAD51C, FANCD2, BARD1, XRCC3, BRCA1, RAD51, and BCCIP (Fig. 3). As BRCA2 plays a major role in DNA repair processes, it becomes evident that harmful mutations in BRCA2 can disrupt not only its own functionality but also that of its interacting partners. This interplay is crucial to understand the broader impact of BRCA2 mutations and their implications in various cellular processes.
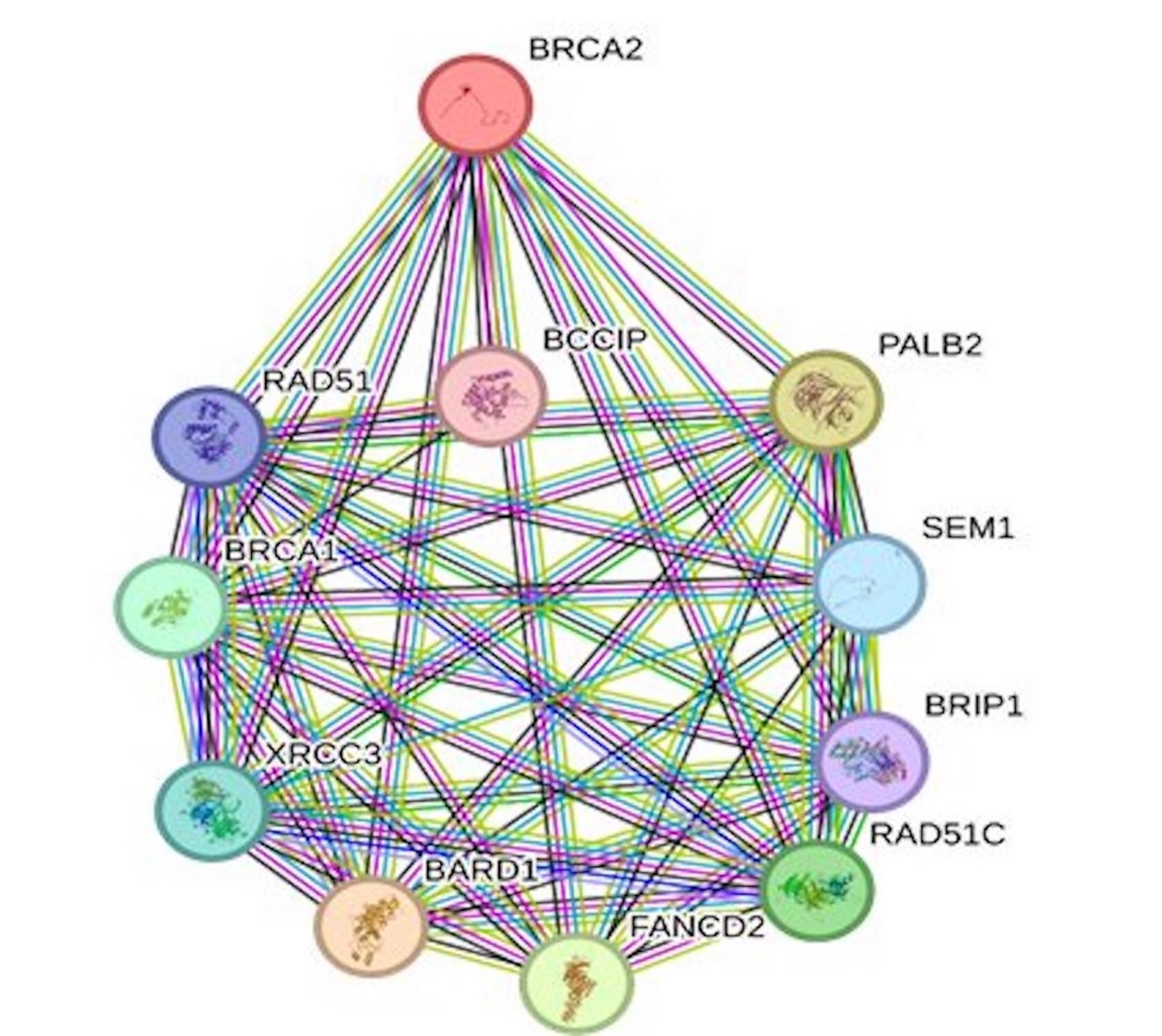
Fig. 3: STRING interaction network of BRCA2 protein showing 10 different interacting proteins.
Identification of a deleterious mutation in the functional domain
InterPro tool was utilized to locate domain regions in BRCA2 protein and to identify the location of deleterious nSNPs in different domains. Utilizing the InterPro database, we identified eight BRC repeat regions and DNA binding domains, comprising the helical domain, OB1, OB2, and OB3 DNA binding domains. Out of the 22 deleterious mutations, a substantial 18 nsSNPs were identified within these DNA binding domains as illustrated in Fig. 4. Additionally, one deleterious mutation was detected in the BRC repeat region. These BRC repeats bind with RAD51, a protein involved in DNA repair and recombination. It is noteworthy that most of these mutations (18) are clustered in the DNA binding region, underscoring its heightened significance. This region’s critical role in DNA repair, given its interaction with DNA, suggests potential implications for the DNA-repairing ability of BRCA2.
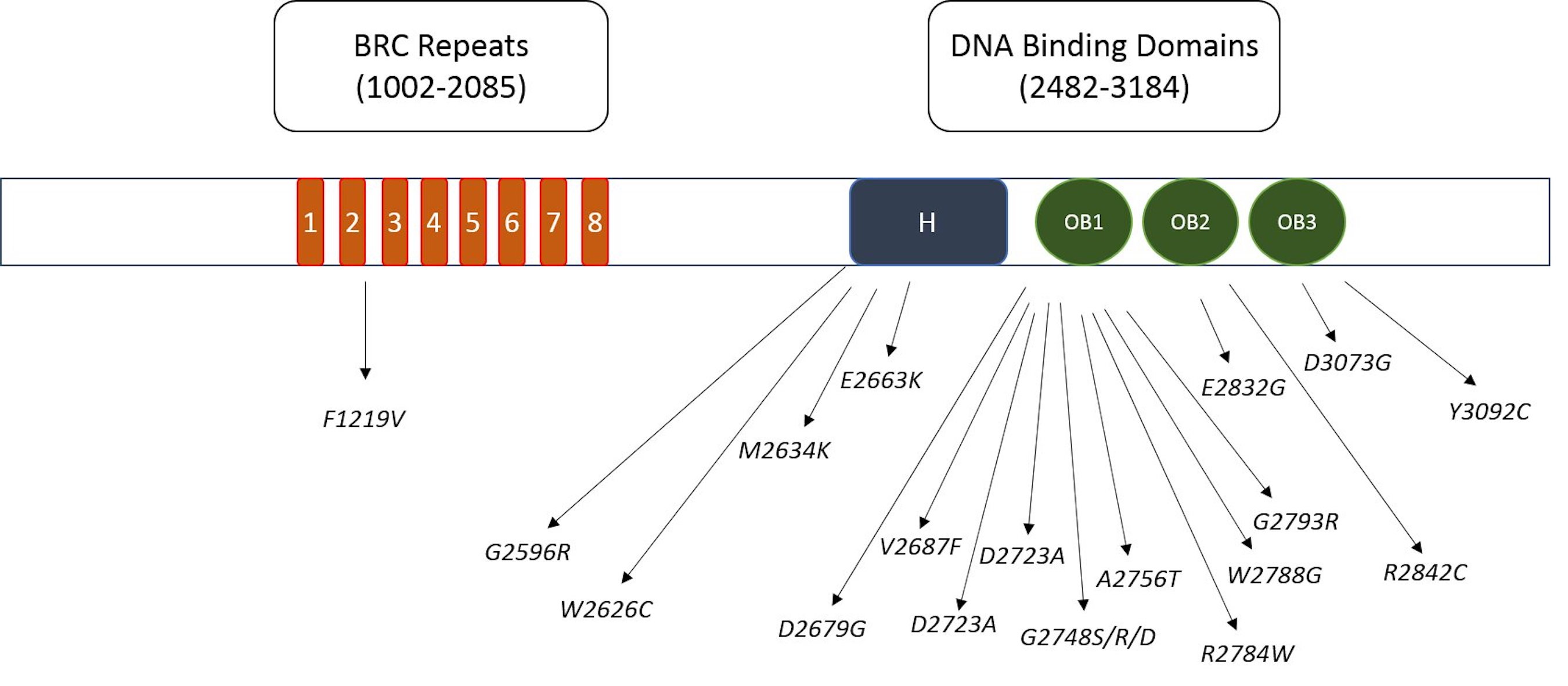
Fig. 4: The schematic representation of identified harmful mutations distributed across the functional domains of BRCA2 protein.
3D Structure Analysis
The 3D structure of the BRCA2 protein was generated using the Swiss model. To ensure comprehensive coverage, we selected a model encompassing amino acids from 2122 to 3373, which notably included 20 out of the deleterious SNPs we had identified. The generated model underwent further refinement through GalaxyRefine and rigorous validation using Errat and PROCHECK. The quality score obtained via Errat was an impressive 92.51%, indicating the reliability of our predicted model. The Ramachandran plot, a crucial measure of the model’s quality, revealed that 88.9% of residues fell within the most favored region, while 6.3% and 1.9% resided in allowed and generously allowed regions, respectively (Fig. 5). These results collectively affirm the satisfactory quality of our BRCA2 3D model. Additionally, mutant protein models were built by manually inserting altered amino acids into the sequence using PyMOL, enriching our analysis of BRCA2 variants (Fig. 6). These models were energy minimized via Swiss PDB viewer and then with Chiron. We extended our analysis by calculating RMSD values for each mutant model. The Root Mean Square Deviation (RMSD) is utilized to calculate the average distance between the α-carbon backbones of wild-type and mutant models. A larger RMSD value indicates a greater deviation of the mutant structure from that of the wild type. The A2231P mutant exhibited the highest RMSD, while the lowest RMSD of 0.034 was observed for the M2643K mutant.
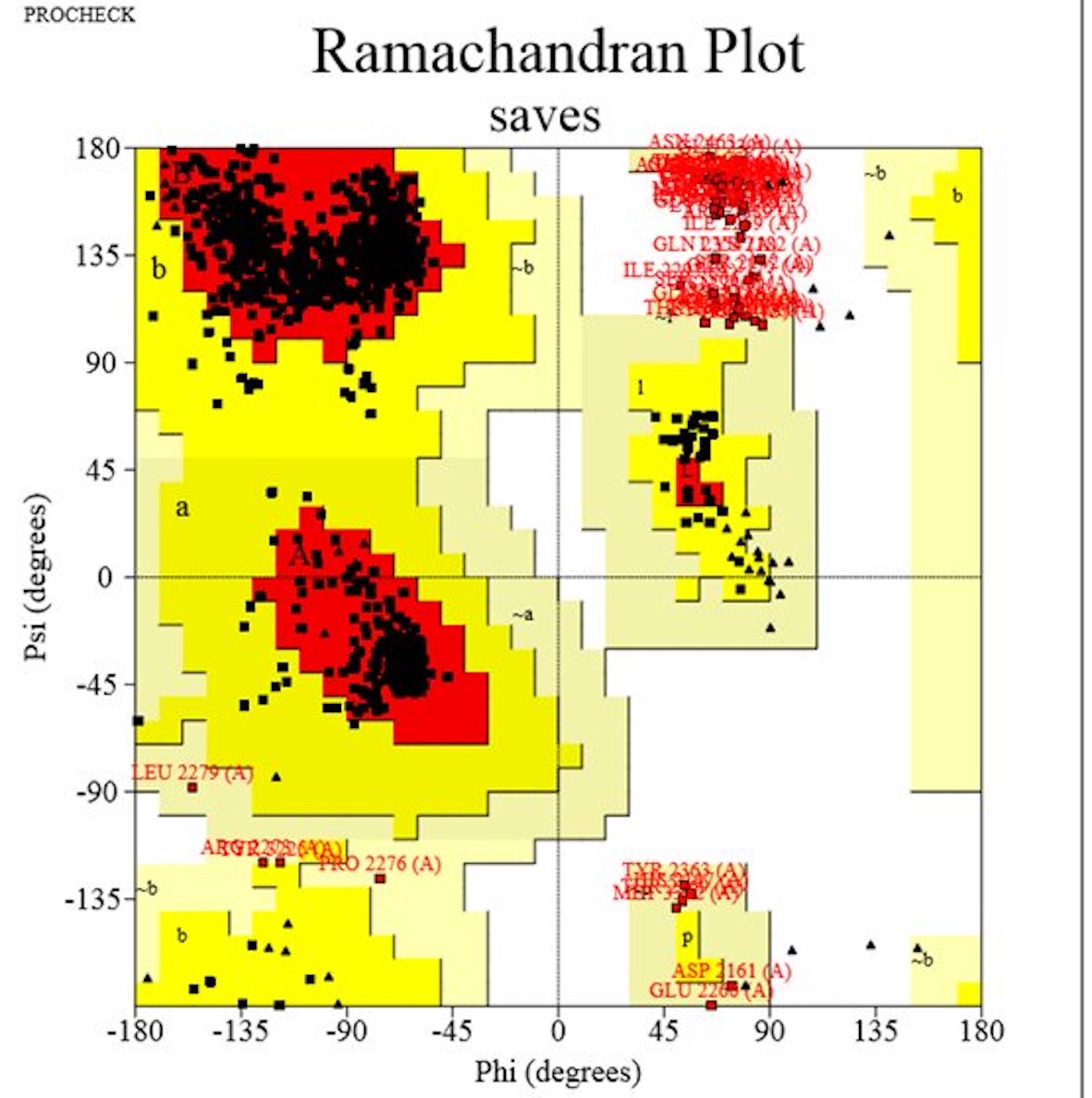
Fig. 5: Ramachandran plot analysis of modeled BRCA2 structure generated by PROCHECK. Residues in most favored regions (A, B, L), Residues in additional allowed regions (a, b, l, p), and residues in generously allowed regions (~a, ~b ~l, ~p).
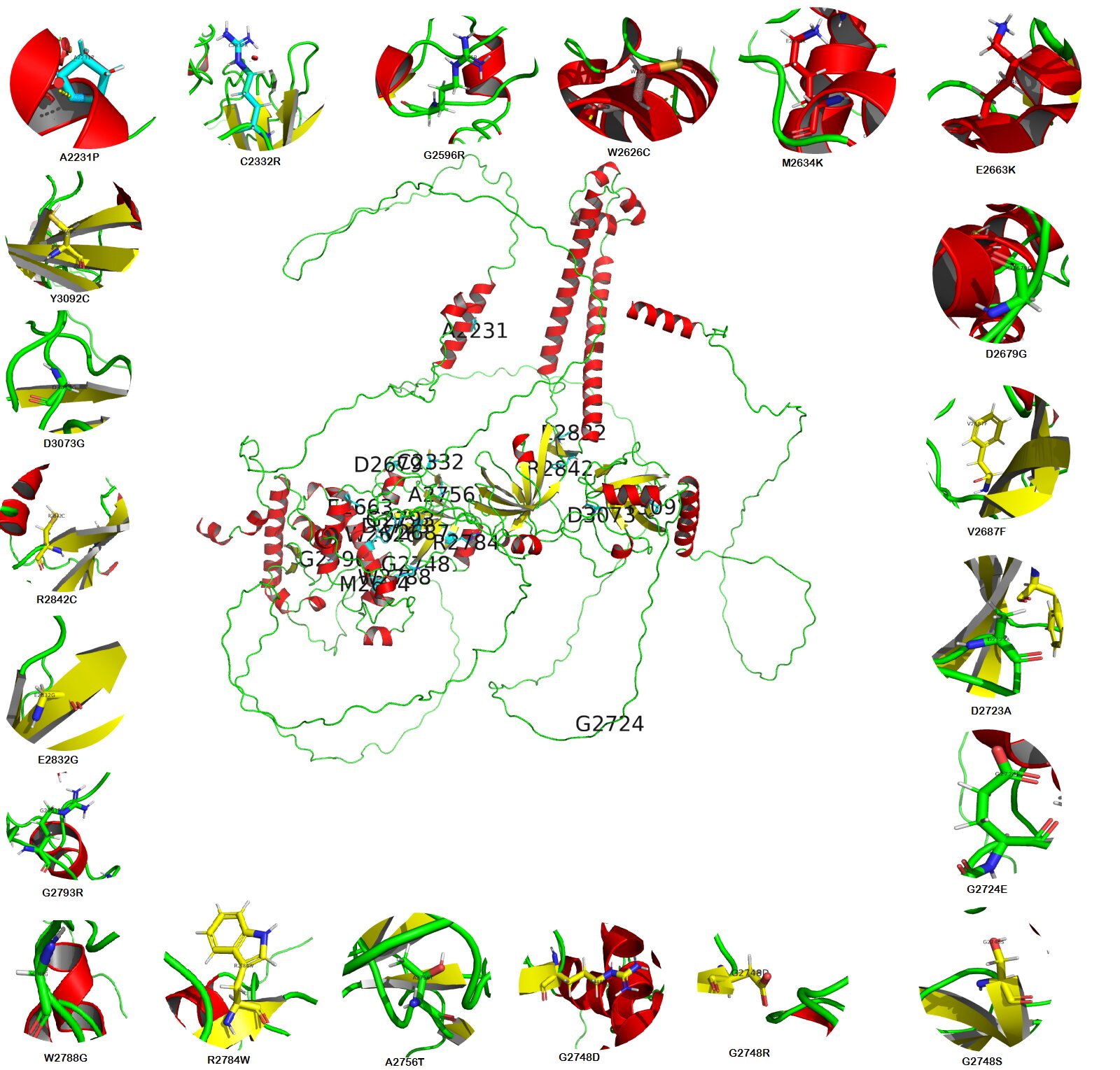
Fig. 6: The central image represents the BRCA2 wild-type model generated by the Swiss Model; Peripheral images showed all (21) missense mutations of BRCA2 protein were generated with PyMol.
Ligand binding site prediction
The coach server was used to predict the ligand binding sites which were then visualized and analyzed using ChimeraX. Using this technique, three ligand-binding sites, site 1 (2513, 2514, 2518, 2520, 252, 2522, 2523, 2526), site 2 (2642, 2645, 2646, 2355, 2659, 2668, 2669, 2670, 2674), site 3 (2725, 2750, 2791, 2792, 2795) (Fig. 7).
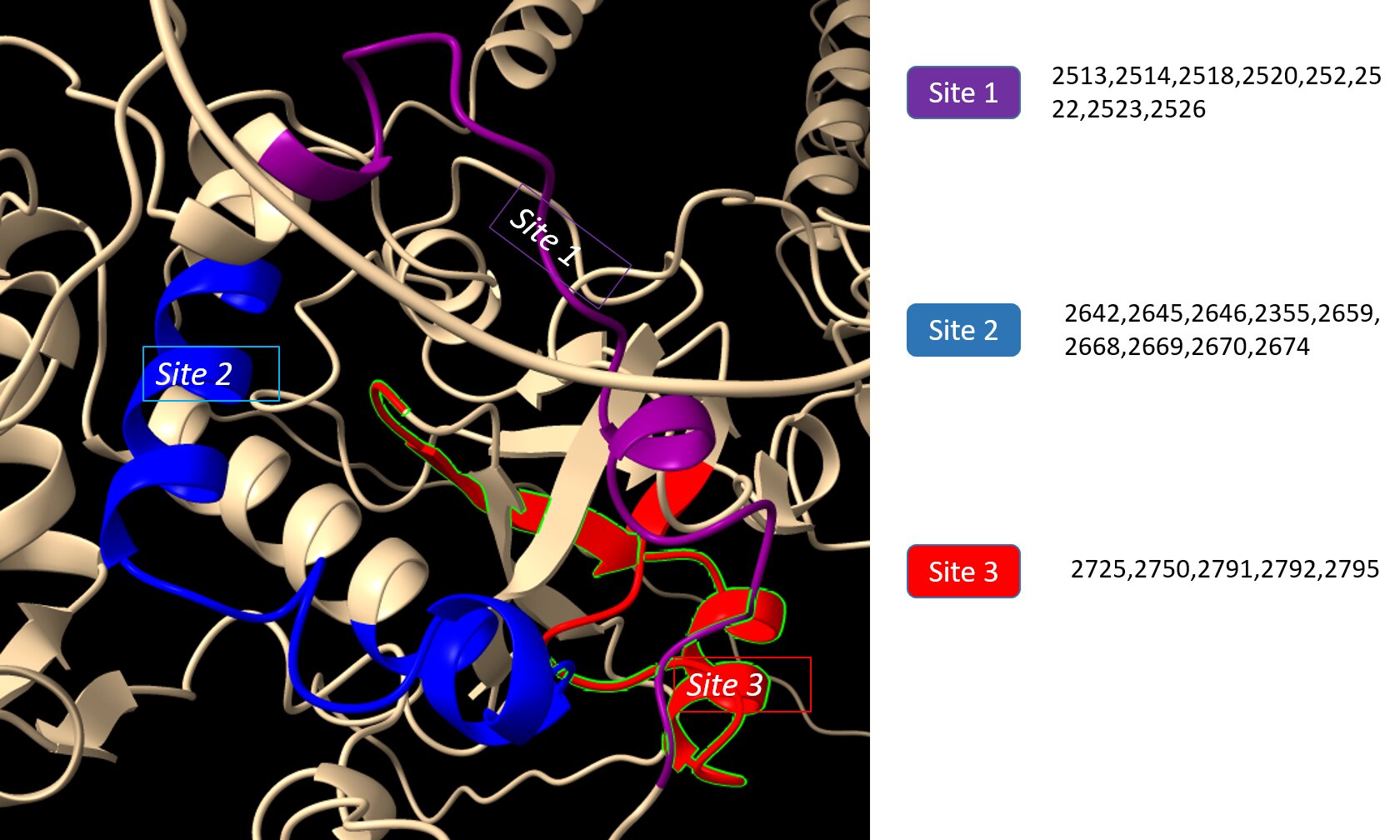
Fig. 7: Ligand binding sites predicted by COACH.
Correlation of BRCA2 Deregulation with Breast and Ovarian Cancer
To comprehend the functional implications of BRCA2 deregulation, we explored the complex relationship between BRCA2 dysregulation and clinical outcomes in cancer patients. Using Kaplan–Meier plot analysis, we discovered distinct repercussions of BRCA2 dysregulation across various cancer types. Our findings indicated that elevated BRCA2 gene expression significantly correlates with reduced survival rates in patients with breast and ovarian cancer (Fig. 8). This emphasizes the major role of BRCA2 in cancer prognosis. A nuanced understanding of how genetic variations, such as SNPs, influence BRCA2 transcriptional regulation and expression could hold immense diagnostic and prognostic potential in disease management.
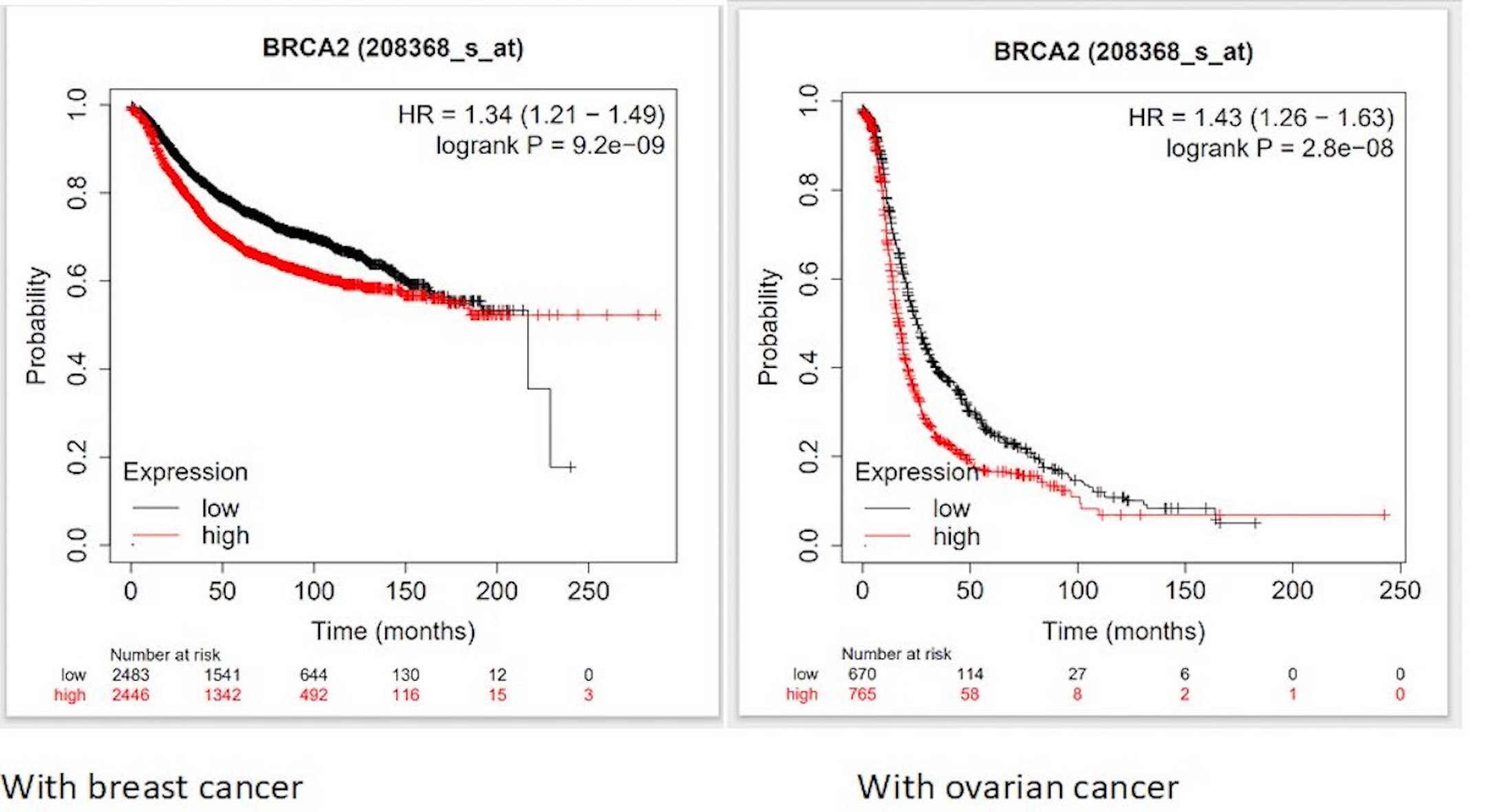
Fig. 8: Kaplan–Meier curves showing the association of BRCA2 expression and survival of patients in (A) Breast, and (B) Ovarian Cancer.
Discussion
Several research studies have identified a connection between variations in the BRCA2 gene and the development of breast cancer [33, 34]. The BRCA2 gene harbors numerous Single Nucleotide Polymorphisms (SNPs) that may influence the susceptibility to breast cancer. We focused on non-synonymous SNPs (nsSNPs) within BRCA2 , aiming to identify the most harmful and deleterious variants that could be linked to the development of breast cancer.
The comprehensive in-silico analysis of BRCA2 SNPs presented in this study provides valuable insights into the potential implications of genetic variations associated with breast cancer. Our exploration began with the retrieval of 7, 921 SNPs from the GenomAD database, with a particular focus on 1, 940 missense or non-synonymous SNPs for further investigation. To assess the deleterious impact of these non-synonymous SNPs, we employed five prediction tools, namely SIFT, PolyPhen, PredictSNP, SNAP2, and PhD-SNP, resulting in the identification of a consensus set of 69 SNPs predicted to be deleterious across all tools. These 69 deleterious SNPs were assessed for their clinical significance by cross-referencing them with ClinVar and VarSome databases. Among the identified SNPs, 16 were classified as pathogenic, indicating a high likelihood of contributing to disease development, while 11 were classified as benign, suggesting they are unlikely to have a significant impact. However, a substantial portion of the SNPs, totaling 42, were categorized as having uncertain significance (VUS). Subsequent MutPred analysis prioritized 48 SNPs based on their potential to damage associated proteins. The MutPred2 scores, coupled with predicted molecular mechanisms, highlighted the significant likelihood of these SNPs influencing molecular processes and contributing to disease pathogenesis. Some of the mutations and their consequences include D2723E, G2724E, A2730P, G2748S, G2748R, G2748D, and A2756T. With a MutPred2 score of 0.638, the D2723E mutation in the BRCA2 gene is expected to result in alterations to metal binding, which may interfere with the coordination of metal ions. Enzymatic activities and structural stability depend on these interactions. Additionally, it is anticipated that the mutation will affect the ordered interface, indicating modifications to structured regions that may affect stability or protein-protein interactions [35]. It is interesting to note that MutPred2 predicts the acquisition of a catalytic site at D2723, suggesting a possible enzymatic activity gain. The observation of the loss of an allosteric site at W2725 raises the possibility of modifications to protein regulation. The complexity of the D2723E mutation and its possible complex effects on BRCA2 function is highlighted by these combined effects. The D2723A mutation, with a higher MutPred2 score of 0.848, alters metal binding like D2723E. However, it predicts a strand gain, which may affect beta-sheet structures and protein stability [36]. This mutation predicts the loss of a catalytic site at D2723, indicating different effects on enzymatic activity than D2723E. The loss of an allosteric site at W2725 suggests that BRCA2 regulatory pathways may change. The G2724E mutation, with a MutPred2 score of 0.838, alters metal binding and predicts strand loss. Beta-sheet structures in the protein may be affected by this change, affecting its stability and function. The MutPred2 score of 0.762 for the A2730P mutation suggests it affects metal binding and ordered interfaces. Gaining a strand and an allosteric site at W2725 suggests structural modifications and new regulatory mechanisms, respectively, highlighting the mutation’s functional complexity. Further categorization of these SNPs revealed associations with altered protein stability, emphasizing their potentially harmful effects. Notably, our study identified 38 SNPs with potential links to cancer through Fathmm analysis, underscoring their relevance in the context of cancer-related molecular mechanisms. Predicting the impact of genomic variations on protein stability, specifically amino acid substitutions, is critical for identifying disease-associated SNPs in proteins [37, 38]. Missense mutations, which alter protein stability, are closely linked to genetic diseases in humans. Certain studies indicate that a decrease in protein stability leads to a rise in protein breakdown, misfolding, and aggregation [39]. This understanding is vital for effective screening of potential disease-causing SNPs in proteins [40]. In our study we calculated stability using I-Mutant and MuPro, revealing 22 SNPs with negative SVM scores, and G values indicative of decreased protein stability. Understanding the secondary structure is essential for comprehending the protein’s folding pattern and its potential interactions with other molecules. Moreover, predictions of BRCA2 secondary structure highlighted the prevalence of random coils, alpha helices, extended strands, and beta turns within the protein’s structure. The protein-protein interaction analysis using the STRING tool identified 10 significant proteins interacting with BRCA2, emphasizing the interconnected nature of cellular processes. Harmful mutations in BRCA2 may not only impact its functionality but also disrupt the network of interacting proteins, particularly those involved in DNA repair processes. The evolutionary conservation analysis using the ConSurf tool demonstrated that all 22 selected SNPs reside within highly conserved regions of the BRCA2 protein. This conservation further supports their functional and structural importance, suggesting their roles in disease development and protein function.
Through the utilization of the InterPro tool, specific domains within the BRCA2 protein were pinpointed, particularly focusing on crucial DNA-binding regions such as the helical domain, OB1, OB2, and OB3. Additionally, the BRCA2 protein encompasses eight BRC repeat regions. Notably, our analysis revealed 18 deleterious mutations (A2231P, C2332R, G2596R, W2626C, M2634K, E2663K, D2679G, V2687F, D2723A, G2724E, G2748S, G2748R, G2748D, A2756T, R2784W, W2788G, G2793R, E2832G, R2842C, D3073G, Y3092C) were identified within the DNA-binding domains, with only one occurring in the BRC repeat region. This underscores the significance of DNA-binding domains as potential hotspots for harmful genetic variations, suggesting a critical role in the protein’s function related to DNA repair and tumor suppression. The association of deleterious mutations predominantly with the DNA-binding domains implies a potential disruption in the protein’s ability to interact with DNA, a fundamental aspect of its role in maintaining genomic stability and facilitating DNA repair. Considering the well-established connection between BRCA2 mutations and an elevated risk of breast cancer, the identification of these mutation-prone domains holds substantial clinical relevance. This insight into the prevalence of deleterious mutations in the DNA-binding regions emphasizes the importance of these domains for researchers, pharmaceuticals, and biotech companies. When developing targeted therapies or diagnostic tools for breast cancer, especially those associated with BRCA2 mutations, careful consideration of these critical DNA-binding domains is paramount. These findings highlight the need for focused attention on the design of interventions to address mutations occurring within these specific regions of the BRCA2 protein. Consequently, drug designers and biotech researchers should take heed of these critical domains when developing targeted therapies or diagnostic tools for breast cancer, particularly those associated with BRCA2 mutations.
The generation of a reliable 3D model of the BRCA2 protein adds a structural dimension to our analysis. The model, refined through GalaxyRefine and validated using Errat and PROCHECK, underscores its quality and reliability. The insertion of altered amino acids into the sequence to create mutant protein models using PyMOL enriches our understanding of specific variants and their potential structural implications. In summary, our In-silico analysis offers a multi-faceted perspective on BRCA2 genetic variations associated with breast cancer. The identification of deleterious mutations, their potential links to cancer, alterations in protein stability, and the structural implications highlighted in the 3D model contribute to a comprehensive understanding of the functional consequences of BRCA2 SNPs. These findings provide a foundation for further experimental validations and clinical investigations, ultimately advancing our knowledge of breast cancer susceptibility and aiding in the development of targeted therapeutic strategies.
Conclusion
In conclusion, our in-silico analysis of BRCA2 genetic variations has revealed a thorough understanding of their potential role in breast cancer susceptibility. The complexity of genetic variations within BRCA2 is demonstrated by the recent identification of 69 deleterious SNPs, including 16 pathogenic variants and 42 of uncertain significance. Certain mutations such as D2723E, G2724E, A2730P, and others were discovered by MutPred analysis, providing insight into their possible impact on molecular processes. In addition, the correlation between these mutations and altered protein stability, as well as their frequent appearance in DNA-binding domains that are highly conserved, emphasize the structural and functional importance of these mutations. The understanding of domains prone to mutation, specifically those linked to DNA-binding regions, is of critical clinical importance and requires concentrated effort in the advancement of targeted therapies and screening techniques for breast cancer. A structural dimension is added to our analysis with the incorporation of a reliable 3D model, which improves our comprehension of variants. Overall, our results offer a strong platform for additional clinical and experimental validations, advancing our knowledge of breast cancer susceptibility and enabling the development of focused treatment approaches.
Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSP2024R191), King Saud University, Riyadh, Saudi Arabia
Authors Declaration
All authors reviewed and authorized the final manuscript and agreed to publish it.
Consent to publish
All the authors have read and approved the article for publication.
Availability of data and materials The manuscript includes all the necessary data; related data may be provided on request from the corresponding author.
Funding
There is no funding received to conduct the study
Authors’ Contributions
HJ did the bioinformatics work and wrote the first draft under the supervision of NUK. MT and AMQ supported the analysis and reviewed the article. NUK, AMQ, MHA and IA critically reviewed the final manuscript.
Disclosure Statement
The authors declared no competing interests and have nothing to disclose.
References
| 1 | Arakelyan A, Melkonyan A, Hakobyan S, Boyarskih U, Simonyan A, Nersisyan L, et al. Transcriptome patterns of brca1-and brca2-mutated breast and ovarian cancers. Int J Mol Sci. 2021;22(3):1266.
https://doi.org/10.3390/ijms22031266 |
| 2 | Gupta P, Dhull AK, Kaushal V, Center R. SYNCHRONOUS VERSUS METACHRONOUS MALIGNANCIES OF BREAST AND OVARY. J. Evid. Based Med. Healthc. 2017;4(25):1486-1488
https://doi.org/10.18410/jebmh/2017/288 |
| 3 | Yoshihara K, Enomoto T, Aoki D, Watanabe Y, Kigawa J, Takeshima N, et al. Association of gBRCA1/2 mutation locations with ovarian cancer risk in Japanese patients from the CHARLOTTE study. Cancer Sci. 2020;111(9):3350-8.
https://doi.org/10.1111/cas.14513 |
| 4 | Zhang L, Wang J, Cui L-Z, Wang K, Yuan M-M, Chen R-R, et al. Successful treatment of refractory lung adenocarcinoma harboring a germline BRCA2 mutation with olaparib: a case report. World J Clin Cases. 2021;9(25):7498.
https://doi.org/10.12998/wjcc.v9.i25.7498 |
| 5 | Toss A, Venturelli M, Molinaro E, Pipitone S, Barbieri E, Marchi I, et al. Hereditary pancreatic cancer: a retrospective single-center study of 5143 Italian families with history of BRCA-related malignancies. Cancers (Basel). 2019;11(2):193.
https://doi.org/10.3390/cancers11020193 |
| 6 | Hsu C-R, Lu T-M, Chin LW, Yang C-C. Possible DNA viral factors of human breast cancer. Cancers (Basel). 2010;2(2):498-512.
https://doi.org/10.3390/cancers2020498 |
| 7 | Sekine M, Nishino K, Enomoto T. Differences in ovarian and other cancers risks by population and BRCA mutation location. Genes (Basel). 2021;12(7):1050.
https://doi.org/10.3390/genes12071050 |
| 8 | Zhang Y. BRCA1, BRCA2 and primary ovarian insufficiency. E3S Web of Conferences. EDP Sciences; 2020; p 05009.
https://doi.org/10.1051/e3sconf/202016505009 |
| 9 | Consortium U. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2010;39(suppl_1):D214-9.
https://doi.org/10.1093/nar/gkq1020 |
| 10 | Flynn RL, Zou L. Oligonucleotide/oligosaccharide-binding fold proteins: a growing family of genome guardians. Crit Rev Biochem Mol Biol. 2010;45(4):266-75.
https://doi.org/10.3109/10409238.2010.488216 |
| 11 | Carreira A, Kowalczykowski SC. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proceedings of the National Academy of Sciences. 2011;108(26):10448-53.
https://doi.org/10.1073/pnas.1106971108 |
| 12 | Le HP, Heyer W-D, Liu J. Guardians of the genome: BRCA2 and its partners. Genes (Basel). 2021;12(8):1229.
https://doi.org/10.3390/genes12081229 |
| 13 | Fenoy IM, Bogado SS, Contreras SM, Gottifredi V, Angel SO. The knowns unknowns: Exploring the homologous recombination repair pathway in Toxoplasma gondii. Front Microbiol. 2016;7:627.
https://doi.org/10.3389/fmicb.2016.00627 |
| 14 | Le HP, Ma X, Vaquero J, Brinkmeyer M, Guo F, Heyer W-D, et al. DSS1 and ssDNA regulate oligomerization of BRCA2. Nucleic Acids Res. 2020;48(14):7818-33.
https://doi.org/10.1093/nar/gkaa555 |
| 15 | Shen L, Zhang S, Wang K, Wang X. Familial Breast Cancer: Disease Related Gene Mutations and Screening Strategies for Chinese Population. Front Oncol. 2021;11:740227.
https://doi.org/10.3389/fonc.2021.740227 |
| 16 | Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248-9.
https://doi.org/10.1038/nmeth0410-248 |
| 17 | Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. 2012
https://doi.org/10.1371/journal.pone.0046688 |
| 18 | Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014;10(1):e1003440.
https://doi.org/10.1371/journal.pcbi.1003440 |
| 19 | Hecht M, Bromberg Y, Rost B. Better prediction of functional effects for sequence variants. BMC Genomics. 2015;16(8):1-12.
https://doi.org/10.1186/1471-2164-16-S8-S1 |
| 20 | Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(D1):D980-5.
https://doi.org/10.1093/nar/gkt1113 |
| 21 | Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(W1):W452-7.
https://doi.org/10.1093/nar/gks539 |
| 22 | Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013;76(1):7-20.
https://doi.org/10.1002/0471142905.hg0720s76 |
| 23 | Capriotti E, Fariselli P. PhD-SNPg: a webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res. 2017;45(W1):W247-52.
https://doi.org/10.1093/nar/gkx369 |
| 24 | Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. 2020;11(1):5918.
https://doi.org/10.1038/s41467-020-19669-x |
| 25 | Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57-65.
https://doi.org/10.1002/humu.22225 |
| 26 | Kircher M, Witten DM, Jain P, O'roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310-5.
https://doi.org/10.1038/ng.2892 |
| 27 | Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(suppl_2):W529-33.
https://doi.org/10.1093/nar/gkq399 |
| 28 | Geourjon C, Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics. 1995;11(6):681-4.
https://doi.org/10.1093/bioinformatics/11.6.681 |
| 29 | Geourjon C, Deleage G. SOPM: a self-optimized method for protein secondary structure prediction. Protein Engineering, Design and Selection. 1994;7(2):157-64.
https://doi.org/10.1093/protein/7.2.157 |
| 30 | Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607-13.
https://doi.org/10.1093/nar/gky1131 |
| 31 | Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236-40.
https://doi.org/10.1093/bioinformatics/btu031 |
| 32 | Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single‐site mutations using support vector machines. Proteins: Structure, Function, and Bioinformatics. 2006;62(4):1125-32.
https://doi.org/10.1002/prot.20810 |
| 33 | Li Q, Guan R, Qiao Y, Liu C, He N, Zhang X, et al. Association between the BRCA2 rs144848 polymorphism and cancer susceptibility: a meta-analysis. Oncotarget. 2017;8(24):39818.
https://doi.org/10.18632/oncotarget.16242 |
| 34 | Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, et al. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst. 1999;91(11):943-9.
https://doi.org/10.1093/jnci/91.11.943 |
| 35 | Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. 2020;11(1):5918.
https://doi.org/10.1038/s41467-020-19669-x |
| 36 | Pejaver V, Mooney SD, Radivojac P. Missense variant pathogenicity predictors generalize well across a range of function‐specific prediction challenges. Hum Mutat. 2017;38(9):1092-108.
https://doi.org/10.1002/humu.23258 |
| 37 | Chuah A, Li S, Do A, Field M, Andrews D. StabilitySort: assessment of protein stability changes on a genome-wide scale to prioritize potentially pathogenic genetic variation. Bioinformatics. 2022;38(17):4220-2.
https://doi.org/10.1093/bioinformatics/btac465 |
| 38 | Gong J, Wang J, Zong X, Ma Z, Xu D. Prediction of protein stability changes upon single-point variant using 3D structure profile. Comput Struct Biotechnol J. 2023;21:354-64.
https://doi.org/10.1016/j.csbj.2022.12.008 |
| 39 | Bross P, Corydon TJ, Andresen BS, Jørgensen MM, Bolund L, Gregersen N. Protein misfolding and degradation in genetic diseases. Hum Mutat. 1999;14(3):186-98.
https://doi.org/10.1002/(SICI)1098-1004(1999)14:3<186::AID-HUMU2>3.0.CO;2-J |
| 40 | Ferrer-Costa C, Orozco M, de la Cruz X. Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J Mol Biol. 2002;315(4):771-86.
https://doi.org/10.1006/jmbi.2001.5255 |