BMI-1 in Breast Cancer – Biological Role and Clinical Implications
Keywords
Abstract
Breast cancer is the most frequent cancer among women. Despite extensive research in recent years the molecular basis of breast cancer development, growth and metastasis remains unclear. Numerous studies highlight the involvement of BMI-1 in tumorigenesis. BMI-1 protein is a key component of Polycomb Repressive Complex 1, which by ubiquitinylation of histone H2A, regulates expression of genes involved in various cellular processes including cell cycle, proliferation and programmed cell death. Overexpression of BMI-1 has been often associated with breast cancer development and progression. This review summarizes the current state of knowledge of BMI-1’s role in breast cancer biology and its potential significance as prognostic marker and potential target of new anticancer therapy.
Introduction
BMI-1 (B-cell-specific Moloney murine leukemia virus integration region) is a key member of the Polycomb Repressive Complex 1. Polycomb group proteins (PcG) are a well-studied group of chromatin modifiers that were originally identified in Drosophila melanogaster [1-3]. Since then, they have been found in other animals, including mammals. Polycomb proteins can be classified into two distinct multiprotein complexes – Polycomb Repressive Complexes 1 and 2 (PRC1 and PRC2) [4]. The complexes have different catalytic activities, but both are associated with transcriptional silencing [5]. Through histone modifications, both Polycomb complexes regulate the expression of factors and genes encoding proteins of signaling pathways thereby affecting numerous molecular processes [6]. Dysregulation of Polycomb repressive complex proteins can lead to abnormal regulation of genes controlling cell division, growth, apoptosis, or migration, leading to cancer development and progression [7].
The PRC1 complex has E3 ubiquitin ligase activity, which in mammal cells specifically modifies histone H2A lysine 119 (H2AK119Ub) and is responsible primarily for its monoubiquitinylation [8]. In recent years, it has become clear that PRC1 is not a single complex, but at least eight different complexes, defined by a set of subunits that confer different biological functions. These complex variants can be classified further as canonical or noncanonical PRC1, depending on whether they contain the CBX (Chromobox) protein or one of two homologous proteins, RYBP (RING1 and YY1 binding protein) and YAF2 (YY1-associated factor 2), respectively [9]. Canonical and noncanonical PRC1 are characterized by the presence of RING1A (Really Interesting New Gene 1A) and its paralog RING1B, which is the core of the PRC1 complex and is responsible for the catalytic activity of PRC1. The PRC1 complexes are numbered PRC1.1 – PRC1.6, depending on which of the six PCGF proteins (Polycomb Group Ring Finger) they contain. The non-canonical variants of PRC1 contain each of six PCGF proteins, but Mel18 (also known as PCGF2) and BMI-1 (also known as PCGF4) are also present in the canonical Polycomb complex [5, 9, 10]. BMI-1, a key component of the PRC1 complex, is considered an oncogene and its overexpression is frequently observed in many types of cancer. BMI-1 plays a crucial role in various cellular processes, including cell cycle regulation, DNA damage response, cell proliferation, aging, apoptosis, and tumorigenesis [11]. In addition, the protein plays an important role in stem cell biology, including cancer stem cells [12].
Characteristics of BMI-1 and its regulation
BMI-1 is encoded by the BMI1 gene located on the short arm of chromosome 10 (10p11.23) and consists of 10 exons and 9 introns. The product is a protein with a molecular weight of about 37 kDa, composed of 326 amino acids. The BMI-1 protein contains 3 characteristic regions in its structure: the central HTH (helix-turn-helix) domain, the N-terminal RING domain, and the C-terminal PEST domain. The RING domain consists of a triple twisted β-sheet, two zinc-binding loops, and an α-helix [13, 14]. It is required for the binding of BMI-1 to the ubiquitin ligase RING1B, which is the catalytic component of the PRC1 complex responsible for the ubiquitinylation of Lys119 of histone H2A. Through the RING domain, BMI-1 can localize within DNA breaks, which is crucial for its role in the DNA Damage Response (DDR) pathway, while the HTH domain facilitates the binding of BMI-1 to the DNA [15]. The PEST domain is a region rich in proline, glutamic acid, serine, and threonine, and its function is to target BMI-1 for degradation [16]. In addition to the three core functional regions, BMI-1 also contains two nuclear localization signals NLS1 and NLS2, but only loss of NLS2 can significantly reduce the nuclear localization of BMI-1 [17].
BMI-1 expression is observed in many tissues and its regulation occurs at the transcriptional, post-transcriptional, and post-translational levels [Fig. 1]. Several transcription factors actively regulate the expression of the BMI1 gene. Transcription factors that affect the direct upregulation of BMI1 expression include MYCN, c-Myc, Twist1, Sp1 and E2F-1. c-Myc and MYCN regulate BMI-1 expression by binding to the E-box sequence of the BMI1 promoter region [18]. Studies by Wang et al. confirmed the function of c-Myc and Sp1 as BMI1 activators and their direct effects on BMI1 both in vitro and in vivo. Furthermore, silencing the expression of c-Myc and Sp1 in the NPSC cell line has the effect of reducing activity [19]. In cancer cells, the increase in BMI1 expression by Twist1 affects the cells' acquisition of stem cell characteristics [20]. The transcription factor involved in the indirect transcriptional upregulation of BMI1 are FOXM1 and SALLA4. FOXM1 indirectly enhances BMI1 expression by increasing c-Myc expression while SALL4 attaches to the BMI1 locus and positively regulates its expression by inducing H3K4 and H3K79 methylation within the promoter region of this gene [21, 22]. Transcription factors that downregulate BMI1 expression are KLF4 and Mel18. Klf4 has been shown to directly bind to the BMI1 promoter and downregulate its expression [23]. Mel-18 downregulates c-Myc expression leading to BMI1 inhibition. Moreover, it was reported that the downregulation of BMI1 through Mel-18 overexpression significantly affects the level of AKT kinase phosphorylation on Ser473 and Thr308 [24].
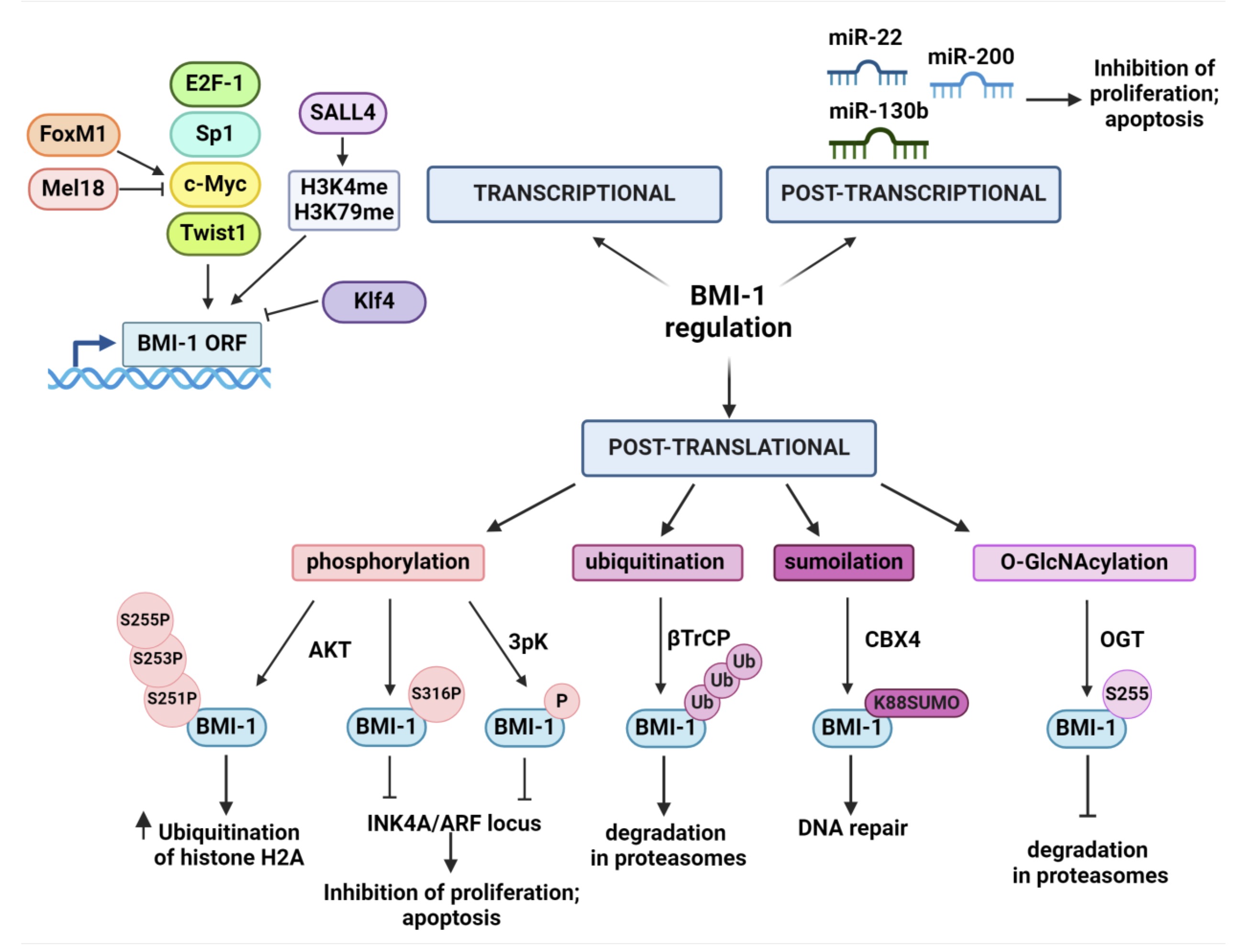
Fig. 1: Regulation of BMI-1 on transcriptional, post-transcriptional and post-translational levels. Several transcription factors are directly or indirectly involved in up- or downregulation the BMI1 gene. The miRNAs are responsible for post-transcriptional regulation of BMI1 influencing the correct regulation of numerous cellular processes. Post-translational modifications (PTMs) of BMI-1 affect its stability and function. Several PTMs that regulate BMI-1’s function have been identified including phosphorylation, ubiquitination, sumoilation and O-GlcNAcylation (detailed description in text). Created with BioRender.com.
BMI1 expression is post-transcriptionally regulated by miRNAs. miRNAs are short single-stranded RNA molecules (21-23 nt) that regulate gene expression through complementarity to the 3'-UTR regions in mRNA. Abnormal expression of miRNAs can affect cellular processes and thus lead to cancer formation and progression. Changes in miRNA expression are associated with the deletion or amplification of the genes encoding them, located at the chromosome break sites [25]. Downregulation of the expression of certain miRNAs that occur frequently in cancer cells can affect the upregulation of BMI1 expression, and therefore play an important role in the regulation of numerous cell processes and the acquisition of stem cell characteristics by cancer cells. Furthermore, miRNAs that regulate the expression of BMI1 can significantly affect tumor sensitivity to radiotherapy or chemotherapy [26]. One of the most important regulators of BMI1 expression in the miR-200 family. MiR-200b and miR-200c are mainly responsible for the inhibition of BMI1 expression in breast, bladder, prostate, head and neck, or melanoma cancer [27 – 31]. In addition, researchers have discovered miRNAs with oncogenic properties that appear to indirectly promote the expression of BMI1. MiR-130b exemplifies a microRNA capable of inducing stem cell-like characteristics in glioma cells. Its overexpression triggers elevated expression of BMI1, a known stem-related regulator, together with pluripotency markers such as CD133, Sox2, Nanog, and c-Myc [32]. In breast cancer cells, miR-22 exhibits an antagonistic effect on tumor suppressor miR-200, leading to an indirect upregulation of BMI1 expression [33].
The function and regulation are critically modulated by post-translational modifications (PTM). These PTMs affect protein stability, localization, and, ultimately, function. MAPKAPK3-mediated phosphorylation of BMI-1 plays a key role during differentiation and development. Interestingly, overexpression of 3pK triggers phosphorylation of BMI-1 and other Polycomb proteins, leading to chromatin dissociation and subsequent activation of INK4A/ARF locus transcription [34]. Furthermore, PI3K/AKT signaling pathway involvement adds another layer of complexity, as AKT-mediated phosphorylation of BMI-1 at specific residues can differentially influence its oncogenic properties. Liu et al. revealed that AKT-mediated phosphorylation of BMI-1 at Ser316 within the PRC1 complex hinders H2A ubiquitination, subsequently preventing its association with the INK4A/ARF locus. This modification impedes BMI-1's ability to promote both tumor growth and cellular proliferation [35]. Interestingly, in mouse prostate cancer cells, phosphorylation of BMI-1 at Ser251, 253, and 255 enhances histone H2A ubiquitination by PRC1, thereby potentiating the protein's oncogenic capacity in an INK4A/ARF-independent manner [36]. This study highlights how residue-specific phosphorylation of BMI-1 differentially modulates its oncogenic properties. The stability and function of the BMI-1 protein are modulated by ubiquitinylation within its PEST domain, mediated by the ubiquitin ligase βTrCP. In MCF-10A cells, βTrCP overexpression promotes BMI-1 degradation in proteasomes. On the contrary, silencing βTrCP expression results in decreased BMI-1 degradation and enhanced pro-oncogenic activity of the protein [37]. Upon DNA damage, the CBX4 protein mediates the attachment of a small protein modifier called SUMO to Lys88 on the BMI-1 protein. This SUMO modification facilitates the recruitment and accumulation of BMI-1 at DNA damage [38]. Ser255 was identified as the specific amino acid residue on the Bmi-1 protein that undergoes O-GlcNAcylation in prostate cancer. This post-translational modification was shown to enhance the stability of the Bmi-1 protein and promote its oncogenic activity, suggesting a potential role in tumorigenesis [39].
BMI-1 in breast cancer biology
BMI-1 affects the expression of genes that encode crucial proteins in numerous biological processes that are disrupted during the development of cancer [12]. BMI-1 overexpression has been associated with various cancers, including breast cancer [40]. In breast cancer cells, BMI-1 has been associated with increased cell proliferation, resistance to apoptosis, and tumor metastasis. It also promotes the formation of cancer stem cells, which are responsible for tumor recurrence and resistance to therapy [41]. The effect of BMI-1 on specific cellular processes in breast cancer will be discussed in the following paragraphs.
BMI-1 in proliferation and cell cycle
Cancer develops from dysregulated cell proliferation, characterized by uncontrolled cell division exceeding normal tissue turnover. Cell cycle play a crucial role in the proliferation of normal cells and its progression is tightly controlled and depends on expression of INK4a/ARF locus which encodes two distinct proteins - p16INK4a and p14Arf that are frequently mutated or deleted in tumors. It has been reported that both p16INK4a and p14Arf expression can be regulated by BMI-1 [42]. In the absence of p16INK4a during the cell cycle, pRB is hyperphosphorylated by the cyclin-dependent kinase complex CDK4 and CDK6. Hyperphosphorylation of pRB results in the failure of this factor to bind and inhibit the transcription factor E2F, resulting in the transcription of genes involved in the transition from G1 to S phase. Inhibition of BMI-1 has been shown to affect p16INK4a expression and prevent CDK4/6 from binding to cyclin D, which affects the activity of this kinase.
p19Arf, through binding to the ubiquitin ligase MDM2, prevents degradation and inactivation of the p53. BMI-1 binding to the p19Arf gene promoter prevents its expression, leading to an increase in MDM2 protein levels. MDM2 leads to p53 degradation, resulting in the inhibition of apoptosis and uncontrolled cell proliferation [43].
The results of many studies indicated that BMI-1 downregulation induce cell cycle arrest and inhibits DNA synthesis in breast cancer cells. Li et al. (2014) observed that BMI-1 silencing in MDA-MB-231 reduced cyclin D1 expression which led to the increase in the number of cells at G0/G1 phase and a reduction in S phase [44]. Xu et al. (2011) showed that BMI-1 downregulation in MCF-7 cells inhibited cell proliferation and induced a decrease of cyclin D1, cyclin E and cdk2 on mRNA and protein levels causing cell cycle arrest [45]. Numerous other studies using specific BMI-1 inhibitor, PTC-209 or RNAi method have proven the participation of BMI-1 in cell cycle regulation and proliferation of different breast cancer cells [41, 46, 47]. Furthermore, the results obtained in in vitro studies were confirmed on an in vivo model using nude mice with tumors induced by injection of MDA-MB-231 cells treated with PTC-209. Tumors with decreased amount of BMI-1 were much smaller and expression of proliferation marker Ki67 was significantly lower compared to the control group [48].
BMI-1 in programmed cell death
Apoptosis, a programmed cell death, plays a crucial role in maintaining cellular homeostasis. In addition to the degradation of redundant cells, apoptosis is also responsible for the elimination of potentially harmful cells, thus inhibiting tumorigenesis. However, cancer cells often acquire the ability to evade apoptosis, which is a hallmark of their uncontrolled growth and proliferation. It has been reported that BMI-1 knockdown induced apoptosis in breast cancer [47, 49]. Moreover, tumors induced in nude mice and treated with BMI-1 siRNA showed increased numbers of apoptotic cells compared with an untreated group and control siRNA group [45]. BMI-1 silencing enhanced the radiosensitivity of MCF-7 cell line and induced apoptosis in cells with elevated p53, p21 and Bax expression and decreased Bcl-2 expression [46]. Another study revealed that miR-630 transfection increased cell apoptosis in BT-549 and HCC1937 cell lines by directly targeting BMI-1. Elevation of cleaved PARP-1, cleaved caspase-3, p21, p27 and pγH2AX was observed in both cell lines [50]. BMI-1 and ABCC5 are target genes of miR-128 which regulates the chemotherapeutic sensitivity of breast cancer cells. Ectopic expression of miR-128 reduced the protein level of BMI-1 and increased apoptosis [51].
Autophagy is an intracellular mechanism where cytoplasmic components are degraded by the lysosome. While autophagy is generally considered a protective mechanism against cancer, its role is more nuanced in the context of breast cancer. On one hand, in the early stages of initiation, autophagy inhibits cancer development by breaking down damaged or harmful components and eventually triggering cell death. On the other hand, in the late stages of tumor development, autophagy is thought to be a mechanism supporting tumor progression and metastasis [52]. Autophagy and BMI-1 have a complex, but still relatively unknown relationship in breast cancer. Griffith et al. (2017) revealed that silencing of BMI1 expression in MDA-MB-231 and SUM159PT breast cancer cell lines, sensitizes cells to ionizing radiation and leads to the activation of autophagy observed by changes of autophagy markers LC3-II and p62. LC3-II plays a crucial role in forming autophagosomes that serve as the main compartments for autophagic cargo, while p62’s primary function in autophagy is to facilitate the delivery of ubiquitinated proteins to autophagic complexes. Silencing BMI1 using shBMI1 and siBMI1 resulted in an increase in LC3-II and a decrease of p62 levels in both cell lines [53]. The other study showed the effect of COPZ1 knockdown, a protein that plays a significant role in regulating vesicle transport within cells, on autophagy induction in MCF-7 cells. COPZ1 knockdown promotes autophagy in breast cancer cells, while BMI-1 overexpression reverses the effect. This suggests that BMI-1 may promote tumorigenesis by counteracting impact of COPZ1 on autophagy [54]. The balance between autophagy and BMI-1 in breast cancer cells may be crucial for determining the overall impact on cancer development and progression. Understanding this intricate relationship is essential for developing more effective cancer treatment.
BMI-1 in cancer progression
The spread of cancer involves two key processes: migration, where tumor cells detach and roam within the tissue, and invasion, where they directly grow and infiltrate nearby healthy tissues. Epithelial-to-mesenchymal transition (EMT) stands as a regulator of both processes. EMT induces a phenotypic switch in epithelial cells, characterized by the downregulation of epithelial markers and the acquisition of mesenchymal characteristics. This phenotypic transformation includes enhanced motility and invasive potential, contributing significantly to various physiological and pathological processes [55]. Overexpression of BMI-1 in breast cancer has a crucial role in the regulation of both migration and invasion by promoting EMT.
The study by Yuan et al. (2015) revealed that increased expression of BMI-1 in Hs578t and MDA-MB-231 breast cancer cells was correlated to increased expression of vimentin, a mesenchymal marker, and decreased expression of E-cadherin, an epithelial marker. Moreover, the link between BMI-1 and the spread of breast cancer cells after ionizing radiation treatment was observed. BMI-1 knockdown abolished the ability of the ionizing radiation to alter the migration of breast cancer cells and the effect of PI3K/AKT signaling inhibition on EMT was blocked. These results suggest that BMI-1 may play significant role in regulation of EMT and migration of breast cancer cells induced by ionizing radiation through activation of PI3K/AKT signaling [56].
The results of several studies showed that BMI-1 silencing leads to a significant reduction in the migration potential of different breast cancer cell types [48, 53, 57]. Interestingly, the mechanisms by which BMI‐1 promotes proliferation and migration of breast cancer cells may depend on cell types. The results of Liu et al. (2023) demonstrate that the in luminal A cells CDKN2D serve as a target gene for BMI‐1, whereas in triple negative cells the BMI‐1/BRCA1 signaling pathway is critical for cell proliferation and migration [48]. It has been found that BMI-1 silencing in breast cancer cells affects expression of EMT markers such as E-cadherin, N-cadherin, vimentin as well as EMT-inducing transcription factors (Snail, Slug, ZEB1 and ZEB2). Moreover, BMI-1 is involved in regulation of expression of matrix metalloproteinases, which are involved in degradation of extracellular matrix proteins and facilitate cell invasion. Depletion of BMI-1 decreased the levels of MMP-2 and MMP-9 secreted by MDA-MB-231 [44].
BMI-1 in breast cancer biology
Cancer stem cells (CSCs) are a subpopulation of tumor cells that share properties with normal tissue stem cells, including self-renewal, differentiation, tumorigenicity, and specific surface markers. CSCs are responsible for the initiation, progression, metastasis, recurrence, and drug resistance becoming a major factor in the failure of conventional therapy and tumor relapse [58]. Cancer stem cells have been found in various types of solid tumors, including breast cancer [59]. Their dysregulated ability to self-renew and differentiation allows them to generate new tumors and spread to other parts of the body, making them a critical target for cancer treatment. A characteristic feature of the BCSCs is the presence of several specific surface markers, including CD44, CD24, ALDH1, CD49f and CD133, often associated with chemotherapy and radiotherapy resistance [60, 61]. Three key signaling pathways, Notch, Wnt and Hedgehog are crucial for self-renewal and differentiation of normal stem cell. Disruption of any of those pathways can transform breast cancer cells into BCSCs [62, 63].
Latest studies indicate the involvement of BMI-1 in the development of BCSCs. The ability of BCSCs to self-renew is indicated by their ability to form mammospheres or tumorspheres. Downregulation of BMI-1 by PTC-209 in mouse breast cancer stem cell line FMMC 419II reduced mammosphere forming potential and also caused cell cycle arrest in vitro. Moreover, decreased BMI-1 expression inhibited tumorigenicity in vivo and lowered expression of BCSCs surface marker CD49f [41]. Sharma et al. (2019) showed that metformin, a well-known drug used in diabetes treatment, significantly decreased the expression of BMI-1 and CD44 in breast cancer cell line MDA-MB-231 and reduced colony and spheroid forming [64].
Transcription factors play a crucial role in cancer development by binding to a particular region of the DNA in the promoters or enhancers of their target genes. Overexpression of transcription factors, especially Oct4, Sox2 and Nanog is often observed in cancer stem cells which is associated with tumor transformation, tumorigenicity, and metastasis [Fig. 2]. These key transcription factors are crucial for maintaining the pluripotency and self-renewal of embryonic stem cells, adult stem cells, and cancer stem cells [65]. The downregulation of those transcription factors results in BCSCs self-renewal inhibition [66 – 69]. BMI-1 also has been identified as a key factor involved in the regulation of self-renewal in both normal and cancer stem cells. Paranjape et al. (2014) demonstrated that BMI-1 regulates the stemness of breast cancer stem cells potentially by modulation of Nanog expression through the NFκB pathway [70].
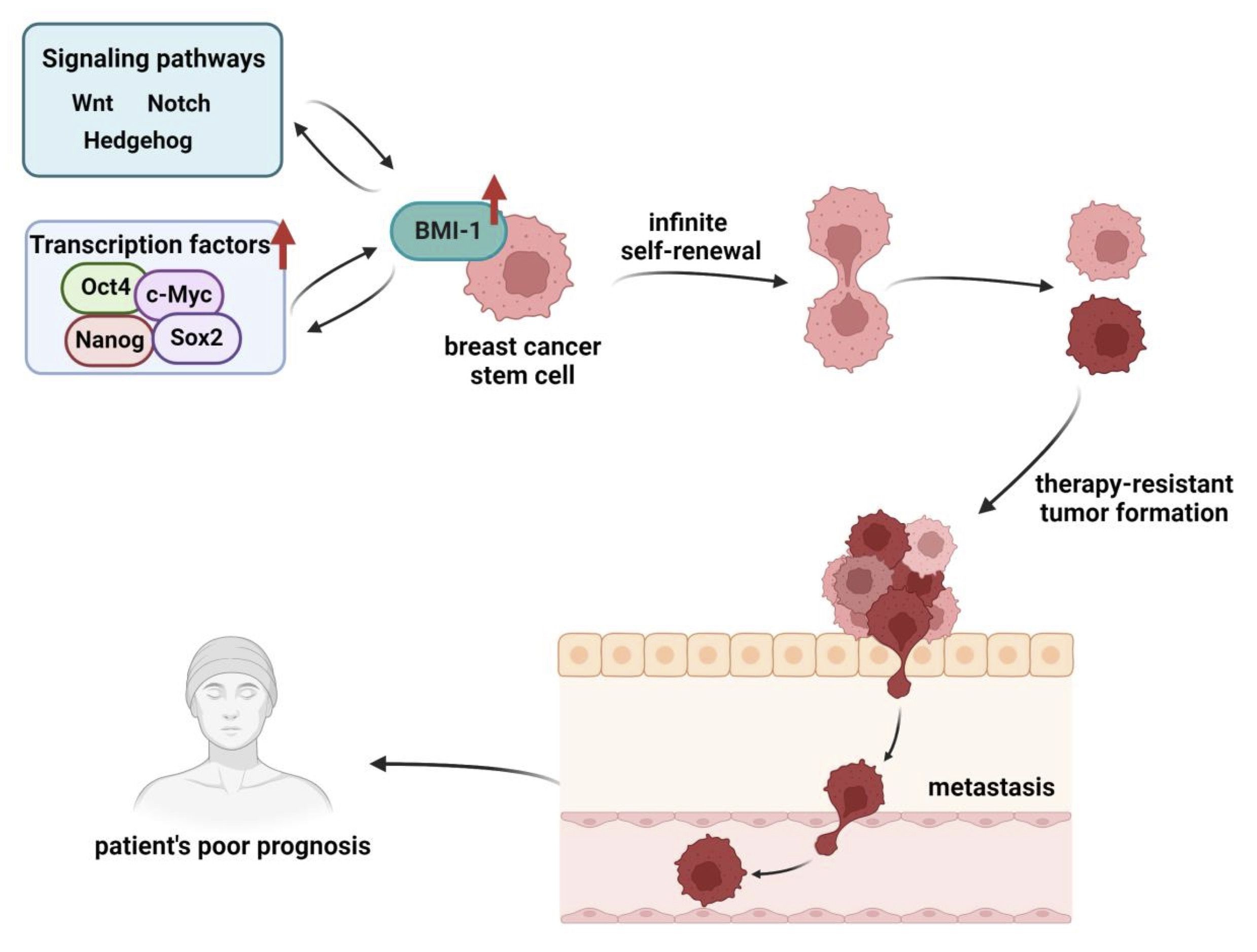
Fig. 2: BMI-1’s involvement and regulation in breast cancer stem cells development and cancer progression. There is an interdependence between BMI-1 and other transcription factors or signaling pathways responsible for transformation of breast cancer cells into BCSCs. Overexpression of transcription factors such as Oct4, Sox2, Nanog, and c-Myc or disturbances in Wnt, Notch, and Hedgehog pathways lead to BMI-1’s overexpression and promotion of self- renewal and maintenance of stem cell-like properties. On the other hand BMI-1 can up-regulate the activity of Wnt and Notch pathways. The changes in expression of BMI-1 may be followed by mutations in genes responsible for therapy-resistant tumor formation and metastasis (detailed description in text). Created with BioRender.com.
There is a growing interest in researching the BMI-1 as well as other epigenetic regulators, including ncRNAs in the context of BCSCs. It was earlier reported that BMI-1 was a target for miR-494-3p in oral squamous cancer cells [71]. Based on that, relationship between this microRNA and BMI1 was researched breast cancer. Chen et al. (2017) showed that treatment with hinokitiol lead to upregulation of miR-494-3p which significantly inhibited BMI-1 expression in mammospheres and suppressed BCSCs self-renewal [72].
Dysfunction of Hedgehog, Wnt and Notch signaling pathways often has been observed in breast cancer [73]. BMI-1 may be directly regulated by these pathways or can influence them in various ways, contributing to the development and progression of breast cancer. It has been found that the Hedgehog signaling pathway promotes self-renewal in mammary stem cells and BCSCs by up-regulating BMI-1. The increased BMI-1 can help mammary stem cells self-renew and maintain their stem cell-like properties [74]. The same interaction between Hedgehog signaling and BMI-1 might also play a role in breast cancer. Treatment of MCF-7 and HCC-1806 cells with dinaciclib inhibited the Hedgehog pathway by decreasing Gli1 expression leading to downregulation of BCSCs markers expression [75]. BMI-1 functions as a positive regulator of the Wnt signaling pathway through the inhibition of Dickkopfs (DKK), a Wnt inhibitor gene. This DKK1 inhibition by BMI-1 leads to the up-regulation of c-Myc, which in turn contributes to a positive feedback loop promoting BMI-1’s own transcriptional activation [76]. BMI-1 also interacts with Notch4, a key factor in Notch signaling and this interaction is required for BMI-1 to promote self-renewal of BCSCs [77].
BMI-1 in breast cancer – clinical implications
Based on the results of numerous pre-clinical studies demonstrating BMI-1’s significant role in tumorigenesis and especially its association with stem cells, it seems that BMI-1 is a good candidate for prognostic marker and even a potential target for the therapy of breast cancer. However, despite evidence of BMI-1 expression deregulation in breast cancer tissues, some controversy exists regarding the impact of BMI-1 overexpression on survival in breast cancer. Pietersen et al. (2008) showed that tumors with high BMI-1 expression display characteristics of less malignant tumors and is correlated with increased survival [78]. Choi et al. (2009) also suggested that BMI-1 expression may be associated with favorable overall survival in breast cancer patients, especially in patients with ER-positive breast cancer [79]. The results of the metaanalysis concerning the prognostic role of high BMI-1 expression in Asian and Caucasian patients with solid tumors showed that in Caucasian populations, high expression of BMI-1 was associated with better overall survival in breast cancer [80]. However, analysis of 252 primary breast cancer cases from Chinese patients performed by Guo et al. (2011) revealed that BMI-1 expression was strongly correlated with larger tumor size lymph node involvement, distant metastasis and advanced clinical stage [81]. More recent studies suggest that there is a correlation between increased BMI-1 expression and clinical progression of breast cancer. Chung et al. (2021) showed that high BMI-1 expression was correlated with poor disease-free survival (DFS) and disease-specific survival (DSS), especially in patients with the luminal subtype of breast cancer. In this study, the machine learning model was used to incorporate BMI-1 to increase the predictive accuracy of the survival rate. The impact of BMI-1 on breast cancer progression seems to depend on the type of breast cancer. A study by Althobiti et al. (2020) demonstrated that high BMI-1 expression was associated with better prognosis in patients with luminal ER-positive breast cancer, but was associated with a poorer survival rate in patients with the triple-negative subtype. In patients with the HER2-positive subtype, there was no significant difference in survival. The results of Chung et al. studies showed that high BMI1 expression was significantly associated with ER and PR negativity. This suggests that BMI-1 plays a complex role in breast cancer and its influence may vary depending on the specific molecular subtypes [82, 83].
While radiation and chemotherapy are widely used to fight breast cancer, tumor resistance, relapse, and metastasis following therapy are major clinical challenges, making treatment less effective. Overexpression of BMI-1 is often linked with treatment resistance. The results of in vitro studies clearly show that inhibition of BMI-1 may cause cancer cells to become more susceptible to treatment thus it can potentially improve the outcomes for cancer patients. It has been reported that expression of BMI-1 was remarkably upregulated in radio- and temozolomide-resistant cells (IRC-R) established from MCF-7 and MDA-MB-231. Silencing of BMI-1 affected cell proliferation of IRC-R cells compared to parental cells suggesting a key role of BMI-1 in radio- and temozolomide- resistance [84]. BMI-1 knockdown increased sensitivity for doxorubicin, one of the most widely used chemotherapeutics, which led to the induction of apoptosis in MCF-7 cell lines and nude mice [46, 49]. It has been reported that BMI-1 overexpression conferred resistance to tamoxifen in two ER+ cell lines MCF-7 and T47D; BMI-1 silencing sensitized the process [85]. Thus, combining BMI-1 inhibitors with standard chemotherapeutic or targeted agents may enhance treatment efficacy and overcome resistance.
One of the most common compounds used to downregulate the expression of BMI-1 in breast cancer cell lines is a small molecule inhibitor PTC-209. This inhibitor functions by reducing the mRNA levels of BMI-1, ultimately leading to decreased protein expression. PTC-209 has shown promising anti-cancer effects in breast cancer cells, including a decrease in cell viability and colony growth. However, PTC-209 has not entered clinical trials because of its limited potency and poor pharmacokinetic properties [86]. PTC-028, has also been characterized as a BMI-1 modulator that has therapeutic potential in ovarian cancer and endometrial cancer [87, 88]. PTC-028 significantly impacts clonal growth and viability of ovarian cancer cells by specifically decreasing BMI-1 through hyper-phosphorylation mediated degradation. It has been found that orally administered PTC-028 exhibits significant, single agent antitumor activity in the orthotopic mouse model of ovarian cancer and causes delayed tumor growth of endometrial cancer compared to the standard-of-care carboplatin and paclitaxel [87, 88]. PTC596 is another small molecule that was identified in a high-throughput small molecule library screen as a potent repressor of BMI-1 in tumor cells, with a favorable safety profile [86]. PTC596 has entered a clinical trial in patients with advanced solid tumors (NCT02404480, NCT03206645, NCT03605550, NCT03761095). Although, PTC596 was originally identified by its ability to inhibit the proliferation of cancer stem cells expressing BMI-1 protein [86], more recent works have demonstrated that the downregulation of BMI-1 protein levels and function is a secondary event to potent mitotic arrest induced by inhibition of tubulin polymerization by PTC596 [89 – 91]. Thus, recent data questioned the significance of BMI-1 for the anti-tumor activity of PTC596 and rather underlined the potent microtubule polymerization inhibition.
In conclusion, without any doubt, the clear link between BMI-1 expression and breast cancer progression exists but the usefulness of BMI-1 as a prognostic factor or potential target for new effective treatment anticancer strategies needs to be proved, and more research is needed to fully understand the role of BMI-1.
Conclusion
Overexpression of BMI-1 found in breast cancer contributes to increased proliferation, metastasis and chemoresistance of cancer cells. High expression of BMI-1 correlated with their progression and poor prognosis. BMI-1 is also one of the most important regulators of the process of differentiation and self-renewal of breast cancer stem cells, which are a major cause of cancer resistance to treatment and recurrence. Studies indicate that direct silencing of BMI-1 expression or reducing its level reduces the ability of CSC to self-renew and initiate tumorigenesis, as well as the metastatic properties and chemoresistance. It is important to note that BMI-1 is still under investigation for breast cancer treatment. More research is needed to determine the best way to target it and ensure the safety and efficacy of such therapies. This includes clinical trials to test the safety and effectiveness of drugs targeting BMI-1 in breast cancer patients.
Acknowledgements
Author Contributions
Conceptualization, A.S. and A.K., writing – original draft preparation, A.S., writing – review and editing, A.K. All authors have read and agreed to the published version of the manuscript.
Funding Sources
This research received no external funding.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Jürgens, G. A group of genes controlling the spatial expression of the bithorax complex in Drosophila. Nature 1985;316, 153-155.
https://doi.org/10.1038/316153a0 |
| 2 | Lewis EB. A gene complex controlling segmentation in Drosophila. Nature 1978;276:565-570.
https://doi.org/10.1038/276565a0 |
| 3 | Simon J. Locking in stable states of gene expression: transcriptional control during Drosophila development. Curr Opin Cell Biol 1995;7:376-385.
https://doi.org/10.1016/0955-0674(95)80093-X |
| 4 | Geng Z, Gao Z. Mammalian PRC1 Complexes: Compositional Complexity and Diverse Molecular Mechanisms. Int J Mol Sci 2020;21:8594.
https://doi.org/10.3390/ijms21228594 |
| 5 | Piunti A, Shilatifard A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat Rev Mol Cell Biol 2021;22:326-345.
https://doi.org/10.1038/s41580-021-00341-1 |
| 6 | Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 2013;20:1147-1155.
https://doi.org/10.1038/nsmb.2669 |
| 7 | Parreno V, Martinez AM, Cavalli G. Mechanisms of Polycomb group protein function in cancer. Cell Res 2022;32:231-253.
https://doi.org/10.1038/s41422-021-00606-6 |
| 8 | Wang H, Wang L, Erdjument-Bromage H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873-878.
https://doi.org/10.1038/nature02985 |
| 9 | Gao Z, Zhang J, Bonasio R, et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 2012;45:344-356.
https://doi.org/10.1016/j.molcel.2012.01.002 |
| 10 | Tavares L, Dimitrova E, Oxley D, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3 [published correction appears in Cell 2012 ;149:1647-1648]. Cell 2012;148:664-678.
https://doi.org/10.1016/j.cell.2011.12.029 |
| 11 | Bhattacharya R, Mustafi SB, Street M, Dey A, Dwivedi SK. Bmi-1: At the crossroads of physiological and pathological biology [published correction appears in Genes Dis. 2015 Dec 08;2(4):353]. Genes Dis 2015;2:225-239.
https://doi.org/10.1016/j.gendis.2015.04.001 |
| 12 | Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells 2012;30:372-378.
https://doi.org/10.1002/stem.1035 |
| 13 | Alkema MJ, Wiegant J, Raap AK, Berns A, van Lohuizen M. Characterization and chromosomal localization of the human proto-oncogene BMI-1. Hum Mol Genet 1993;2:1597-1603.
https://doi.org/10.1093/hmg/2.10.1597 |
| 14 | Li Z, Cao R, Wang M, Myers MP, Zhang Y, Xu RM. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J Biol Chem 2006;281:20643-20649.
https://doi.org/10.1074/jbc.M602461200 |
| 15 | Ginjala V, Nacerddine K, Kulkarni A, et al. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol 2011;31:1972-1982.
https://doi.org/10.1128/MCB.00981-10 |
| 16 | Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci 1996;21:267-271.
https://doi.org/10.1016/S0968-0004(96)10031-1 |
| 17 | Cohen KJ, Hanna JS, Prescott JE, Dang CV. Transformation by the Bmi-1 oncoprotein correlates with its subnuclear localization but not its transcriptional suppression activity. Mol Cell Biol 1996;16:5527-5535.
https://doi.org/10.1128/MCB.16.10.5527 |
| 18 | Huang R, Cheung NK, Vider J, et al. MYCN and MYC regulate tumor proliferation and tumorigenesis directly through BMI1 in human neuroblastomas. FASEB J 2011;25:4138-4149.
https://doi.org/10.1096/fj.11-185033 |
| 19 | Wang HB, Liu GH, Zhang H, et al. Sp1 and c-Myc regulate transcription of BMI1 in nasopharyngeal carcinoma. FEBS J. 2013;280:2929-2944.
https://doi.org/10.1111/febs.12299 |
| 20 | Wu KJ, Yang MH. Epithelial-mesenchymal transition and cancer stemness: the Twist1-Bmi1 connection. Biosci Rep 2011;31:449-455.
https://doi.org/10.1042/BSR20100114 |
| 21 | Li SK, Smith DK, Leung WY, et al. FoxM1c counteracts oxidative stress-induced senescence and stimulates Bmi-1 expression. J Biol Chem 2008;283:16545-16553.
https://doi.org/10.1074/jbc.M709604200 |
| 22 | Yang J, Chai L, Liu F, et al. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci U S A 2007;104:10494-10499.
https://doi.org/10.1073/pnas.0704001104 |
| 23 | Yu T, Chen X, Zhang W, et al. Regulation of the potential marker for intestinal cells, Bmi1, by β-catenin and the zinc finger protein KLF4: implications for colon cancer. J Biol Chem 2012;287:3760-3768.
https://doi.org/10.1074/jbc.M111.316349 |
| 24 | Guo WJ, Zeng MS, Yadav A, et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res 2007;67:5083-5089.
https://doi.org/10.1158/0008-5472.CAN-06-4368 |
| 25 | Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther 2016;1:15004.
https://doi.org/10.1038/sigtrans.2015.4 |
| 26 | Sugihara H, Ishimoto T, Watanabe M, et al. Identification of miR-30e* regulation of Bmi1 expression mediated by tumor-associated macrophages in gastrointestinal cancer. PLoS One 2013;8(11):e81839.
https://doi.org/10.1371/journal.pone.0081839 |
| 27 | Polytarchou C, Iliopoulos D, Struhl K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proc Natl Acad Sci U S A 2012;109:14470-14475.
https://doi.org/10.1073/pnas.1212811109 |
| 28 | Martínez-Fernández M, Dueñas M, Feber A, et al. A Polycomb-mir200 loop regulates clinical outcome in bladder cancer.
|
| 29 | Yu J, Lu Y, Cui D, et al. miR-200b suppresses cell proliferation, migration and enhances chemosensitivity in prostate cancer by regulating Bmi-1. Oncol Rep 2014;31:910-918.
https://doi.org/10.3892/or.2013.2897 |
| 30 | Lo WL, Yu CC, Chiou GY, et al. MicroRNA-200c attenuates tumour growth and metastasis of presumptive head and neck squamous cell carcinoma stem cells. J Pathol 2011;223:482-495.
https://doi.org/10.1002/path.2826 |
| 31 | Liu S, Tetzlaff MT, Cui R, Xu X. miR-200c inhibits melanoma progression and drug resistance through down-regulation of BMI-1. Am J Pathol 2012;181:1823-1835.
https://doi.org/10.1016/j.ajpath.2012.07.009 |
| 32 | Zhu G, Wang Y, Mijiti M, Wang Z, Wu PF, Jiafu D. Upregulation of miR-130b enhances stem cell-like phenotype in glioblastoma by inactivating the Hippo signaling pathway. Biochem Biophys Res Commun 2015;465:194-199.
https://doi.org/10.1016/j.bbrc.2015.07.149 |
| 33 | Song SJ, Poliseno L, Song MS, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013;154.:311-324.
https://doi.org/10.1016/j.cell.2013.06.026 |
| 34 | Voncken JW, Niessen H, Neufeld B, et al. MAPKAP kinase 3pK phosphorylates and regulates chromatin association of the polycomb group protein Bmi1. J Biol Chem 2005;280:5178-5187.
https://doi.org/10.1074/jbc.M407155200 |
| 35 | Liu Y, Liu F, Yu H, et al. Akt phosphorylates the transcriptional repressor bmi1 to block its effects on the tumor-suppressing ink4a-arf locus. Sci Signal 2012;5:ra77.
https://doi.org/10.1126/scisignal.2003199 |
| 36 | Nacerddine K, Beaudry JB, Ginjala V, et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin Invest 2012;122:1920-1932.
https://doi.org/10.1172/JCI57477 |
| 37 | Sahasrabuddhe AA, Dimri M, Bommi PV, Dimri GP. βTrCP regulates BMI1 protein turnover via ubiquitination and degradation. Cell Cycle 2011;10:1322-1330.
https://doi.org/10.4161/cc.10.8.15372 |
| 38 | Ismail IH, Gagné JP, Caron MC, et al. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res 2012;40:5497-5510.
https://doi.org/10.1093/nar/gks222 |
| 39 | Li Y, Wang L, Liu J, et al. O-GlcNAcylation modulates Bmi-1 protein stability and potential oncogenic function in prostate cancer. Oncogene 2017;36:6293-6305.
https://doi.org/10.1038/onc.2017.223 |
| 40 | Wang Y, Zhe H, Ding Z, Gao P, Zhang N, Li G. Cancer stem cell marker Bmi-1 expression is associated with basal-like phenotype and poor survival in breast cancer. World J Surg 2012;36:1189-1194.
https://doi.org/10.1007/s00268-012-1514-3 |
| 41 | Srinivasan M, Bharali DJ, Sudha T, et al. Downregulation of Bmi1 in breast cancer stem cells suppresses tumor growth and proliferation. Oncotarget 2017;8:38731-38742.
https://doi.org/10.18632/oncotarget.16317 |
| 42 | Chiba T, Seki A, Aoki R, et al. Bmi1 promotes hepatic stem cell expansion and tumorigenicity in both Ink4a/Arf-dependent and -independent manners in mice. Hepatology 2010;52:1111-1123.
https://doi.org/10.1002/hep.23793 |
| 43 | Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999;397:164-168 .
https://doi.org/10.1038/16476 |
| 44 | Li H, Song F, Chen X, Li Y, Fan J, Wu X. Bmi-1 regulates epithelial-to-mesenchymal transition to promote migration and invasion of breast cancer cells. Int J Clin Exp Pathol 2014;7:3057-3064.
|
| 45 | Xu Z, Liu H, Lv X, Liu Y, Li S, Li H. Knockdown of the Bmi-1 oncogene inhibits cell proliferation and induces cell apoptosis and is involved in the decrease of Akt phosphorylation in the human breast carcinoma cell line MCF-7. Oncol Rep 2011;25:409-418.
https://doi.org/10.3892/or.2010.1078 |
| 46 | Liu ZG, Liu L, Xu LH, et al. Bmi-1 induces radioresistance in MCF-7 mammary carcinoma cells. Oncol Rep 2012;27:1116-1122.
https://doi.org/10.3892/or.2011.1615 |
| 47 | Wu XM, Liu X, Bu YQ, et al. RNAi-mediated silencing of the Bmi-1 gene causes growth inhibition and enhances doxorubicin-induced apoptosis in MCF-7 cells. Genet Mol Biol 2009;32:697-703.
https://doi.org/10.1590/S1415-47572009005000092 |
| 48 | Liu JY, Jiang YN, Huang H, et al. BMI-1 promotes breast cancer proliferation and metastasis through different mechanisms in different subtypes. Cancer Sci 2023;114:449-462.
https://doi.org/10.1111/cas.15623 |
| 49 | Wu X, Liu X, Sengupta J, et al. Silencing of Bmi-1 gene by RNA interference enhances sensitivity to doxorubicin in breast cancer cells. Indian J Exp Biol 2011;49:105-112.
|
| 50 | Gong XF, Yu AL, Tang J, et al. MicroRNA-630 inhibits breast cancer progression by directly targeting BMI1. Exp Cell Res 2018;362:378-385.
https://doi.org/10.1016/j.yexcr.2017.11.039 |
| 51 | Zhu Y, Yu F, Jiao Y, et al. Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5 [published correction appears in Clin Cancer Res. 2023 Jul 14;29(14):2738]. Clin Cancer Res 2011;17:7105-7115.
https://doi.org/10.1158/1078-0432.CCR-11-0071 |
| 52 | Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer 2017;17:528-542.
https://doi.org/10.1038/nrc.2017.53 |
| 53 | Griffith J, Andrade D, Mehta M, et al. Silencing BMI1 radiosensitizes human breast cancer cells by inducing DNA damage and autophagy. Oncol Rep 2017;37:2382-2390.
https://doi.org/10.3892/or.2017.5478 |
| 54 | Chen S, Li H, Chen S, Wang B, Zhang K. BMI1 promotes the proliferation and inhibits autophagy of breast cancer cells by activating COPZ1. Clin Transl Oncol 2022;24:2166-2174.
https://doi.org/10.1007/s12094-022-02869-w |
| 55 | Son H, Moon A. Epithelial-mesenchymal Transition and Cell Invasion. Toxicol Res 2010;26:245-252.
https://doi.org/10.5487/TR.2010.26.4.245 |
| 56 | Yuan W, Yuan Y, Zhang T, Wu S. Role of Bmi-1 in regulation of ionizing irradiation-induced epithelial-mesenchymal transition and migration of breast cancer cells. PLoS One 2015;10:e0118799.
https://doi.org/10.1371/journal.pone.0118799 |
| 57 | Sulaiman S, Arafat K, Iratni R, Attoub S. PTC-209 Anti-Cancer Effects Involved the Inhibition of STAT3 Phosphorylation. Front Pharmacol 2019;10:1199.
https://doi.org/10.3389/fphar.2019.01199 |
| 58 | Crabtree JS, Miele L. Breast Cancer Stem Cells. Biomedicines 2018;6:77.
https://doi.org/10.3390/biomedicines6030077 |
| 59 | Ghasemi F, Sarabi PZ, Athari SS, Esmaeilzadeh A. Therapeutics strategies against cancer stem cell in breast cancer. Int J Biochem Cell Biol 2019;109:76-81.
https://doi.org/10.1016/j.biocel.2019.01.015 |
| 60 | Butti R, Gunasekaran VP, Kumar TVS, Banerjee P, Kundu GC. Breast cancer stem cells: Biology and therapeutic implications. Int J Biochem Cell Biol 2019;107:38-52.
https://doi.org/10.1016/j.biocel.2018.12.001 |
| 61 | de Beça FF, Caetano P, Gerhard R, et al. Cancer stem cells markers CD44, CD24 and ALDH1 in breast cancer special histological types. J Clin Pathol 2013;66:187-191.
https://doi.org/10.1136/jclinpath-2012-201169 |
| 62 | Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol 2012;6:620-636.
https://doi.org/10.1016/j.molonc.2012.10.006 |
| 63 | Bozorgi A, Khazaei M, Khazaei MR. New Findings on Breast Cancer Stem Cells: A Review. J Breast Cancer. 2015;18(4):303-312 doi:10.4048/jbc.2015.18.4.303
https://doi.org/10.4048/jbc.2015.18.4.303 |
| 64 | Sharma A, Bandyopadhayaya S, Chowdhury K, et al. Metformin exhibited anticancer activity by lowering cellular cholesterol content in breast cancer cells. PLoS One 2019;14:e0209435.
https://doi.org/10.1371/journal.pone.0209435 |
| 65 | Yang L, Shi P, Zhao G, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther 2020;5:8.
https://doi.org/10.1038/s41392-020-0110-5 |
| 66 | Sun Y, Wang Y, Fan C, et al. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol Cancer 2014;13:137.
https://doi.org/10.1186/1476-4598-13-137 |
| 67 | Morata-Tarifa C, Jiménez G, García MA, et al. Low adherent cancer cell subpopulations are enriched in tumorigenic and metastatic epithelial-to-mesenchymal transition-induced cancer stem-like cells. Sci Rep 2016;6:18772.
https://doi.org/10.1038/srep18772 |
| 68 | Oh J, Yoon HJ, Jang JH, Kim DH, Surh YJ. The standardized Korean Red Ginseng extract and its ingredient ginsenoside Rg3 inhibit manifestation of breast cancer stem cell-like properties through modulation of self-renewal signaling. J Ginseng Res 2019;43:421-430.
https://doi.org/10.1016/j.jgr.2018.05.004 |
| 69 | Yang F, Cui P, Lu Y, Zhang X. Requirement of the transcription factor YB-1 for maintaining the stemness of cancer stem cells and reverting differentiated cancer cells into cancer stem cells. Stem Cell Res Ther 2019;10:233.
https://doi.org/10.1186/s13287-019-1360-4 |
| 70 | Paranjape AN, Balaji SA, Mandal T, et al. Bmi1 regulates self-renewal and epithelial to mesenchymal transition in breast cancer cells through Nanog. BMC Cancer 2014;14:785.
https://doi.org/10.1186/1471-2407-14-785 |
| 71 | Weng JH, Yu CC, Lee YC, Lin CW, Chang WW, Kuo YL. miR-494-3p Induces Cellular Senescence and Enhances Radiosensitivity in Human Oral Squamous Carcinoma Cells. Int J Mol Sci 2016;17:1092.
https://doi.org/10.3390/ijms17071092 |
| 72 | Chen SM, Wang BY, Lee CH, et al. Hinokitiol up-regulates miR-494-3p to suppress BMI1 expression and inhibits self-renewal of breast cancer stem/progenitor cells. Oncotarget 2017;8:76057-76068.
https://doi.org/10.18632/oncotarget.18648 |
| 73 | Zardawi SJ, O'Toole SA, Sutherland RL, Musgrove EA. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol Histopathol 2009;24:385-398.
|
| 74 | Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006;66:6063-6071.
https://doi.org/10.1158/0008-5472.CAN-06-0054 |
| 75 | Tsao AN, Chuang YS, Lin YC, Su Y, Chao TC. Hedgehog signaling and Bmi-1 regulate self-renewal of norcer cells by targeting the FoxM1 and Hedgehog signaling pathway. Oncol Rep 2022;47:105.
https://doi.org/10.3892/or.2022.8316 |
| 76 | Cho JH, Dimri M, Dimri GP. A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J Biol Chem 2013;288:3406-3418.
https://doi.org/10.1074/jbc.M112.422931 |
| 77 | Kim SH, Singh SV. Mammary cancer chemoprevention by withaferin A is accompanied by in vivo suppression of self-renewal of cancer stem cells. Cancer Prev Res (Phila) 2014;7:738-747.
https://doi.org/10.1158/1940-6207.CAPR-13-0445 |
| 78 | Pietersen AM, Horlings HM, Hauptmann M, et al. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res 2008;10:R109.
https://doi.org/10.1186/bcr2214 |
| 79 | Choi YJ, Choi YL, Cho EY, et al. Expression of Bmi-1 protein in tumor tissues is associated with favorable prognosis in breast cancer patients. Breast Cancer Res Treat 2009;113:83-93.
https://doi.org/10.1007/s10549-008-9909-4 |
| 80 | Shao Y, Geng Y, Gu W, Ning Z, Jiang J, Pei H. Prognostic role of high Bmi-1 expression in Asian and Caucasian patients with solid tumors: a meta-analysis. Biomed Pharmacother 2014;68:969-977.
https://doi.org/10.1016/j.biopha.2014.10.017 |
| 81 | Guo BH, Feng Y, Zhang R, et al. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer 2011;10:10.
https://doi.org/10.1186/1476-4598-10-10 |
| 82 | Chung Y, Min KW, Kim DH, et al. High BMI1 Expression with Low CD8+ and CD4+ T Cell Activity Could Promote Breast Cancer Cell Survival: A Machine Learning Approach. J Pers Med 2021;11:739.
https://doi.org/10.3390/jpm11080739 |
| 83 | Althobiti M, Muftah AA, Aleskandarany MA, et al. The prognostic significance of BMI1 expression in invasive breast cancer is dependent on its molecular subtypes. Breast Cancer Res Treat 2020;182:581-589.
https://doi.org/10.1007/s10549-020-05719-x |
| 84 | Yan Y, Wang Y, Zhao P, Ma W, Hu Z, Zhang K. BMI-1 Promotes Self-Renewal of Radio- and Temozolomide (TMZ)-Resistant Breast Cancer Cells. Reprod Sci 2017;24:1620-1629.
https://doi.org/10.1177/1933719117697255 |
| 85 | Ojo D, Lin X, Wu Y, Cockburn J, Bane A, Tang D. Polycomb complex protein BMI1 confers resistance to tamoxifen in estrogen receptor positive breast cancer. Cancer Lett 2018;426:4-13.
https://doi.org/10.1016/j.canlet.2018.03.048 |
| 86 | Nishida Y, Maeda A, Kim MJ, et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J 2017;7:e527.
https://doi.org/10.1038/bcj.2017.8 |
| 87 | Dey A, Xiong X, Crim A, et al. Evaluating the Mechanism and Therapeutic Potential of PTC-028, a Novel Inhibitor of BMI-1 Function in Ovarian Cancer. Mol Cancer Ther 2018;17:39-49.
https://doi.org/10.1158/1535-7163.MCT-17-0574 |
| 88 | Buechel M, Dey A, Dwivedi SKD, et al. Inhibition of BMI1, a Therapeutic Approach in Endometrial Cancer. Mol Cancer Ther 2018;17:2136-2143.
https://doi.org/10.1158/1535-7163.MCT-17-1192 |
| 89 | Eberle-Singh JA, Sagalovskiy I, Maurer HC, et al. Effective Delivery of a Microtubule Polymerization Inhibitor Synergizes with Standard Regimens in Models of Pancreatic Ductal Adenocarcinoma. Clin Cancer Res 2019;25:5548-5560.
https://doi.org/10.1158/1078-0432.CCR-18-3281 |
| 90 | Bolomsky A, Muller J, Stangelberger K, et al. The anti-mitotic agents PTC-028 and PTC596 display potent activity in pre-clinical models of multiple myeloma but challenge the role of BMI-1 as an essential tumour gene. Br J Haematol 2020;190:877-890.
https://doi.org/10.1111/bjh.16595 |
| 91 | Jernigan F, Branstrom A, Baird JD, et al. Preclinical and Early Clinical Development of PTC596, a Novel Small-Molecule Tubulin-Binding Agent. Mol Cancer Ther 2021;20:1846-1857.
https://doi.org/10.1158/1535-7163.MCT-20-0774 |