Original Article - DOI:10.33594/000000756
Accepted 28 December 2024 - Published online 31 January 2025
Optimized Protocol For DNA Extraction from Human Whole Blood
Keywords
Abstract
Background/Aims:
DNA isolation is the initial process in genetic research. The product is used in many PCR reactions (PCR-RFLP, Real-Time PCR, multiplex PCR). That is why it is important to optimize DNA isolation protocol to obtain a good quality of DNA. Our first attempts at isolation, conducted using Purification Kit, did not result in sufficient concentration (6.414 ng*μL-1) and purity (A-260/280) of 0.764 of isolated DNA.Methods:
We used twice the recommended amount of tissue and cell lysis solution to get more effective cell lysis. We extend the time of vortexing, centrifugation and incubation at critical steps. We manipulated the speed and temperatures of centrifugation. We used cold iso-propanol to get white strands of DNA faster. When rinsing with ethanol we used cold alcohol. We tested efficiency of two methods of drying of ethanol to achieve optimal DNA pureness. We leave the isolated DNA for 20 minutes to evaporate the ethanol and then resuspend nucleic acid in TE Buffer.Results:
Our modifications resulted in the improvement of isolation efficiency. After optimization we achieved DNA concentration (in range of 50-150 ng*μL-1) and purity (A 260/280) of 1.735. Similar results for DNA parameters were achieved from the whole blood frozen for 2-3 months (concentration in the range of 125.762 ng*μL-1, pureness: 1.761) and from blood frozen for 18 months (117.94 ng*μL-1 and 1.7194, respectively). We performed electrophoresis after each isolation to confirm the effectiveness of optimized procedure. The refinements we used in DNA isolation are more efficient than those recommended in DNA Purification Kits.Conclusion:
Our results confirm that optimized DNA protocol fulfills the conditions of good extraction technique: it is relatively fast and easy to perform yet it guarantees a high reproducibility, specificity and sensitivity. There are also no dangerous or harmful steps. Our paper demonstrates innovative and effective approach. It confirms a high effectiveness of method regardless of duration of sample freezing, as well as introduce important modifications (timing, temperature conditions, drying details, absence of K-proteinase) that make overall procedure more productive and relatively fast.Introduction
The previous research shows that DNA amplification by polymerase chain reaction (PCR) have been routinely used to analyze sequence differences in medical diagnosis of diseases, and population studies [1, 2, 3, 4, 5]. RFLP and molecular sequencing have been extensively used to track the phylogeny of different species including human using deoxyribonucleic acid (DNA) fragment amplified through PCR. DNA digestion by restriction endonuclease is recommended before PCR amplification in case of circular and high molecular mass of DNA. The restriction digestion before PCR amplification of large number of mtDNA samples sometimes gives problems due to a high rate of sequence evolution of such DNA. Such phenomena in mtDNA may introduce extra site of restriction enzyme used to linearize DNA in samples in the region of amplification and consequently desired amplification is not achieved [1, 3].
A good quality of DNA is required for many molecular analyses making isolation of DNA one of the most basic procedures performed in a molecular laboratory. The Swiss physician and biologist Friedrich Miescher was the first scientist to extract DNA in 1869 [6, 7]. Later Meselson and Stahl (1958) [8] developed a laboratory procedure for DNA isolation [6, 8]. Today we have many different protocols and commercially available DNA Purification Kits specialized in extracting DNA from a wide variety of biological materials [9]. The recommended protocols normally involve four basic steps: 1) cell lysis followed by 2) denaturation of nucleoproteins, then 3) removal of contaminants, and finally 4) DNA precipitation [9, 10]. DNA isolation methods are divided into two main categories: solution-based DNA extraction and solid-phase DNA extraction [10].
Rather outdated technique of DNA extraction was based on the density gradient centrifugation in the presence of cesium chloride/ethidium bromide. Separation of DNA with satisfying efficiency took place as a result of differential density between cesium ion and water with intercalation of ethidium bromide (in this way there was a possibility to obtain separation of each DNA as individual bands). The method was highly labor absorbing and time-consuming. Furthermore, it exacted expensive laboratory equipment and working with toxic ethidium bromide [11, 12].
Phenol-chloroform is the classic organic liquid-liquid extraction procedure that separates mixtures of molecules based on differential solubility [9]. In this method cell lysis is achieved by sodium dodecyl sulfate (SDS), followed by denaturation of nucleoproteins, removal of contaminants (by phenol and phenol-chloroform), and then DNA precipitation with isopropanol [9, 13, 10]. In the salting out method proteinase K or laundry powder is used to inactivate enzymes, while a high sodium chloride concentration is used to purify the DNA [14, 13, 10]. Both of these solution-based DNA extractions are still in common use. A modified salting out method was developed into a commercial kit, e.g. Epicentre Master Pure™ DNA Purification Kit.
Solid-phase DNA extraction is faster, easier, more efficient and less problematic than traditional methods, which is why it is commonly used in commercial kits [14, 10]. Generally, after cell lysis, DNA is bound to solid phase, then contaminants are washed off and finally DNA is diluted. All steps usually require centrifugal force, using different pH and buffer conditions. Silica matrices are the most popular solid phase [9, 10]. Today there are modification of membranes, which allows to isolate DNA more efficiency [15, 16]. The silica particles are positively charged and thus have a high affinity for negatively charged DNA molecules. In case of silica based solid phase columns chaotropic buffer often plays the role of protein denaturant as well as cofactor stimulating adsorption of nucleic acids. The key feature of chaotropic ions – enlarged charge delocalization – gives them a potential of deranging neighboring hydrogen bonding which favors protein solubility in the water [17]. One more critical factor is pH which implicates that final effectiveness of the isolation depends on the interaction between DNA, silica and chaotropic molecules as a function of pH. Taking it into consideration the choice of miniaturized silica columns with small pores may act profitably bringing higher effectiveness of adsorption and elution for dilute nucleic acids yet these columns could also be vulnerable to plugging or negative disruptions from proteins, carbohydrates or lipids surface passivation [10, 17].
Another popular method employs magnetic beads made of magnetite or maghemite coated with silica. After DNA is attached to the magnetic pellet it is immobilized to remove supernatant, and then washed with alcohol. Nucleic acids are then suspended in buffer and the beads are eluted [18, 9, 10]. Therefore, the technique of isolation that uses chemically or biologically modified magnetic particles can be considered as altered version of solid phase extraction. Accordingly adjusted nanoparticles can effectively separate cells, ions, pollutants, proteins as well as DNA. Separation is based on binding of specific targets on the surface of magnetic particles. Advantages of this technique arise from micro- and nanoparticles conveniences: relatively low price, versatility, recyclability and compatibility with various biological solutions. There are different types of nanoparticles varied in specification and efficiency of binding. Among them are silica, carbon, silver or gold nanoparticles occasionally with specifically modified surfaces (e.g. the addition of oligonucleotides or amino-coated nanoparticles) [19, 11].
The role of such modifications is to improve binding properties as well as to stabilize particles. Furthermore, size, charge or chemical composition of particles influence on such features as magnetization potential, density or porosity which appear fundamental for effectiveness of binding biomolecules. The surface of particle may also shape its general stability and change the value of zeta potential. Finally, ionic strength and pH of environment as well as the conformation of DNA jointly influence on binding potential of DNA to particles [20]. Therefore, coating the surface of magnetic particle is essential. E.g., binding of nucleic acids to silica with chaotropic salts is widely applied in extraction of nucleic acids from various sample matrices into solutions that are free from inhibitors. In this approach mobility of magnetic particles is an advantage enabling transport of adsorbed biomolecules into relatively small volumes which produce increment in the concentration [21].
Another positive is fast and easy separation of particles bound with biomolecules in the presence of magnetic field (permanent magnet). Thus, in this method nucleic acids are less susceptible for degradation (which in traditional isolation may appear as a consequence of centrifugation). Magnetic separation also permits very simple elimination of potential inhibitors of PCR reaction (e.g. phenolic compounds or polysaccharides) [22]. Finally, relatively wide range of commercially available magnetic carriers (obtained for example from biopolymers or inorganic magnetic materials) made this technique broadly accessible. Despite diversified range of particles diameters – in general larger irregular and possibly porous surface guarantee the most effective binding of nucleic acids (optimally spherical beads of the uniform size) [22, 11].
Anion exchange resins are substances with positively charged di-ethyl-aminoethyl cellulose (DEAE-cellulose) groups on their surfaces. DEAE can react with negatively charged nucleic acids [9, 10]. DNA is deposited on the membrane during the electrophoresis and is more difficult to elute [23]. The development of faster, more effective, relatively low-cost yet stable and repetitive techniques of isolation is a matter of unabated interest. One of the most promising issue is the use of ionic liquids which are liquid molten salts (at the temperature less than 100oC) consist of large unsymmetrical organic cations and organic or inorganic anions [24, 25].
Among certain advantages of ionic liquids are their stability, only tiny volatility and high solvation capability for the broad range of compounds. They also may serve as surface-active ingredients that largely improve the extraction results. Finally, ionic liquids can be involved in and modulate many types of extractions like liquid-liquid (for instance based on the hydrophobic ionic liquids or aqueous biphasic systems), solid-liquid or solid-phase isolations (with IL-modified materials) [24, 25]. What is more, ionic liquids may be potentially applied in the technology of storage of DNA. Serious disadvantage of a long term storage of DNA is a risk of increased oxidative or structural damages especially due to the freeze-thaw of samples. On the other hand, keeping of DNA in room temperature often requires dehydration conditions which may disturb its conformation (exchange of B-form of DNA to A-form) or cause denaturation [26, 25]. Ionic liquids can act as stabilizers of DNA physiological conformation maintaining structural and chemical properties of B-form even for a longer periods of time. Furthermore, ionic liquids provoke conformational transitions in DNA in order to retain B conformation. This phenomenon prompts to use ionic liquids as components of DNA storage media [26, 27]. However, there are also some disadvantages connected with possible ionic liquids toxicity. E.g., it can enter into aquatic environments and remain there for a long time. In this situation high stability of ionic liquids seriously reduce their biodegradability and create a threat for aquatic organisms. On the enzymatic level ionic liquids show inhibitory activities which may lead among others to perturbations of important neurological processes. Ionic liquids toxicity is probably associated with cationic moiety and side-chain length [25].
In modern days genetic studies are popular in research laboratories. Many regions in DNA are being studied for their influence on the development of diseases [3]. It is important to collect and storage samples to conduct many studies to observe influence of changes in DNA on the development diseases. In some studies, the methods may not be satisfactory, therefore it is advisable to retain samples for future studies using newer, more reliable methods [28]. To study one DNA sample for multiple disease predispositions we need effective DNA isolation method (which results with high concentration and good purify and storage nucleic acid) or secure several samples of material from one patient (and isolate DNA when will be performed analysis). Storage of isolated nucleic acid is expensive procedure. There are over 100 companies in the world which perform isolation and long-term DNA storage. Unfortunately, this procedure costs in the range from a hundred to a thousand of United States dollar [28, 29, 30]. Commercially available extraction kits usually deliver pure, double-stranded DNA – the frequently accompanying disadvantage may be significant DNA loss. This problem is especially augmented when DNA undergoes extraction from dried blood spots which is often connected with lower (even 10-fold) DNA recovery rate compared to extraction from the whole blood. It is also worth mention that the limit of detection tend to be lower for extraction from whole blood (better sensitivity) when compared to the dried blood spots [5]. However, the cost per extraction is important and the usage of commercial kits may be connected with expenses but it is rather comfortable when many samples need to be processed [5, 31].
In this paper we discuss DNA isolation using the Epicentre Master Pure™ DNA Purification Kit. The aim of our study was to optimize the isolation of DNA from human peripheral blood using the Master Pure™ DNA Purification Kit in order to obtain 90-100% performance. Isolated DNA using the method described in the original protocol is occasional for analyses, but not for our research, where we used the PCR-RFLP method. That is why we have undertaken to optimize the DNA isolation method. Besides, the original protocol is useful mainly for the isolation of the fresh blood DNA. However, due to the fact that in most cases, frozen blood is used for scientific research, we have adapted the method to our material for such needs. Thanks to this, it will broaden the scope of application of the standard set. Therefore, our optimized method proposed here will allow to isolate DNA from frozen blood. The novel modification details we used were not specified in the protocol instructions from the manufacturer which led us to develop the optimization process outlined in this paper. A further aim of this work was to develop a DNA isolation protocol for use in both in a long-term (about 18 month of storage) and a short-term frozen blood (2-3 months of storage).
Materials and Methods
Peripheral blood was collected from the ulnar vein from patients hospitalized in the Department of Eye Diseases, Nicolaus Copernicus University Hospital No. 1 named of Dr. A. Jurasz in Bydgoszcz, Poland. Patients filled in the declarations of informed and voluntary consent for their participation in the research in the scope of this work.
We used frozen human whole blood for optimization of the protocol of DNA isolation. Samples were collected in vials with K3EDTA, which is common anticoagulant in laboratory diagnostics. Both samples (short-term 2-3 months and long-term approximately 18 months) were frozen. Long-term frozen blood, from 50 patients, was stored for 18 months at –80oC while short-term frozen blood from 50 patients was stored at –80oC for 2-3 months. The same kit (the same component), the same solution bottles, etc. were used for all compared samples, which guaranteed equal conditions for the DNA isolation process. This is important to allow for a proper comparison.
We used an Illumina Epicentre Master Pure™ DNA Purification Kit for isolation [19]. The kit consists of Red Cell Lysis Solution (120 mL), Tissue and Cell Lysis Solution (60 mL), 2X T and C Lysis Solution (50 mL), MPC Protein Precipitation Reagent (55 mL), RNase A (5 μg*μL-1 400 μL), Proteinase K (50 μg*μL-1 200 μL), TE Buffer (17 mL (10 mM Tris-HCl, 1 mM EDTA). One Master Pure™ DNA Purification Kit is enough to perform 200 DNA purifications [32]. We also used iso-propanol and 70% ethanol from the Sigma company.
Additional equipment used in the optimization methods included a centrifuge (Eppendorf), a laboratory incubator (Poll CL-60), a vortex (Eppendorf), a Nanodrop (Thermo Scientific) and a pipette with variable capacity (Eppendorf).
Our isolation protocol was divided into 2 parts:
- ) lysis, where we lysed erythrocytes, leukocytes and digested RNA.
- ) precipitation of total DNA, including removal of proteins, precipitation of DNA and dissolution of the nucleic acid in TE buffer. The manufacturer’s protocol for our sample is as follows [26]: ‘B2. DNA Purification for 200 μL of Whole Blood (with RBC Lysis).
Expected yield: 3-9 μg of DNA (see [33]):
- Draw 5 mL of blood into an EDTA Vacutainer tube. Transfer 200 μL of whole blood into a micro-centrifuge tube. As it is commonly known, multiple research are based on the frozen blood, because it is hard to collect fresh blood from multiple patients during a long-period time. That is why we should test the possibility to isolate good quality DNA from long-term frozen blood.
- Add 600 μL of Red Cell Lysis Solution. Invert three times to mix and then flick the bottom of the tube to suspend any remaining material.
- Incubate at room temperature for 5 minutes and then vortex briefly. Continue incubating at room temperature for an additional 5 minutes, followed again by brief vortexing.
- Pellet the white blood cells by centrifugation for 25 seconds in a micro-centrifuge.
- Remove most of the supernatant, leaving approximately 25 μL of liquid. Vortex to suspend the pellet.
- Resuspend the white blood cells in 300 μL of Tissue and Cell Lysis Solution by pipetting several times.
- Add 1 μL of RNase A and mix thoroughly.
- Incubate at 37°C for 30 minutes.
- Place the samples on ice for 3-5 minutes and then proceed to precipitation of total DNA.
- Add 175 μL of MPC Protein Precipitation Reagent to 300 μL of lysed sample and vortex vigorously for 10 seconds.
- Pellet the debris by centrifugation for 10 minutes at ≥10.000 x g in a microcentrifuge.
- Transfer the supernatant to a clean centrifuge tube and discard the pellet.
- Add 500 μL of iso-propanol to the recovered supernatant. Mix by inverting the tube 30-40 times.
- Pellet the DNA by centrifugation at 4°C for 10 minutes in a micro-centrifuge.
- Carefully pour off the iso-propanol without dislodging the DNA pellet.
- Rinse twice with 70% ethanol, being careful to not dislodge the pellet. Centrifuge briefly if the pellet is dislodged. Remove all of the residual ethanol with a pipette.
- Resuspend the DNA in 35 μL of TE Buffer. Quantitate the DNA by electrophoresis, spectrophotometry, or fluorimetry.
Our revised protocol differed in the following areas:
We transferred the supernatant to a clean microcentrifuge tube with a press stud (1.5 mL; these equipment are safe-lock tubes.
We added an additional step in our protocol at this point.
We added an additional step in our protocol at this point.
We added an additional step in our protocol at this point:
Statistical analysis was performed with STATISTICA v.13 (TIBCO Software Inc. 2017) and Microsoft Excel v.2019. Wilcoxon test was conducted with the confidence level α=0.05 and obtained p-values below 0.05 were recognized as statistically significant while p-values that topped 0.05 were recognized as statistically insignificant. Wilcoxon test is designed to compare results for the same samples obtained in various conditions. By using this test is possible to compare results of DNA concentration in the same sample before and after optimization, and the impact of procedure or sample storage on final DNA concentration.
Measurements of DNA quality parameters (concentration and pureness) were accomplished with Nano-Drop 2000 (Thermo Scientific). Application of 2 µL of DNA suspended in TE Buffer was sufficient for effective analysis. Electrophoresis was performed with horizontal electrophoresis apparatus (MS Major Science MP-300V) while results were documented with photo chamber (Syn-gene G:BOX ChemiXR5). Illumina Epicentre Master Pure™ DNA Purification Kit [32] (Cat. No MCD85201) was selected to perform isolation procedure based on detailed product specification – compatible with our needs – as well as favorable experiences from previous researches conducted in our laboratory with this specific purification kit. All procedures and all stages of optimization was performed with the same purification kit with original reagents provided by manufacturer. Introduction of each modification of original procedure was followed by accurate and multiple examination of after-effects and evaluation of positive impact on DNA concentration and purity. Final results of optimization are presented and accordingly discussed in succeeding section of this paper.
Results
Our first attempts at DNA isolation – conducted strictly adhering to the manufacturer’s protocol did not result in sufficient DNA concentration (ng*μL-1) or purity (A 260/280). The parameters shows, that samples were useless for PCR reactions. Descriptive statistics for these results are presented in Table 1 and Figures 1-3 (n=50).
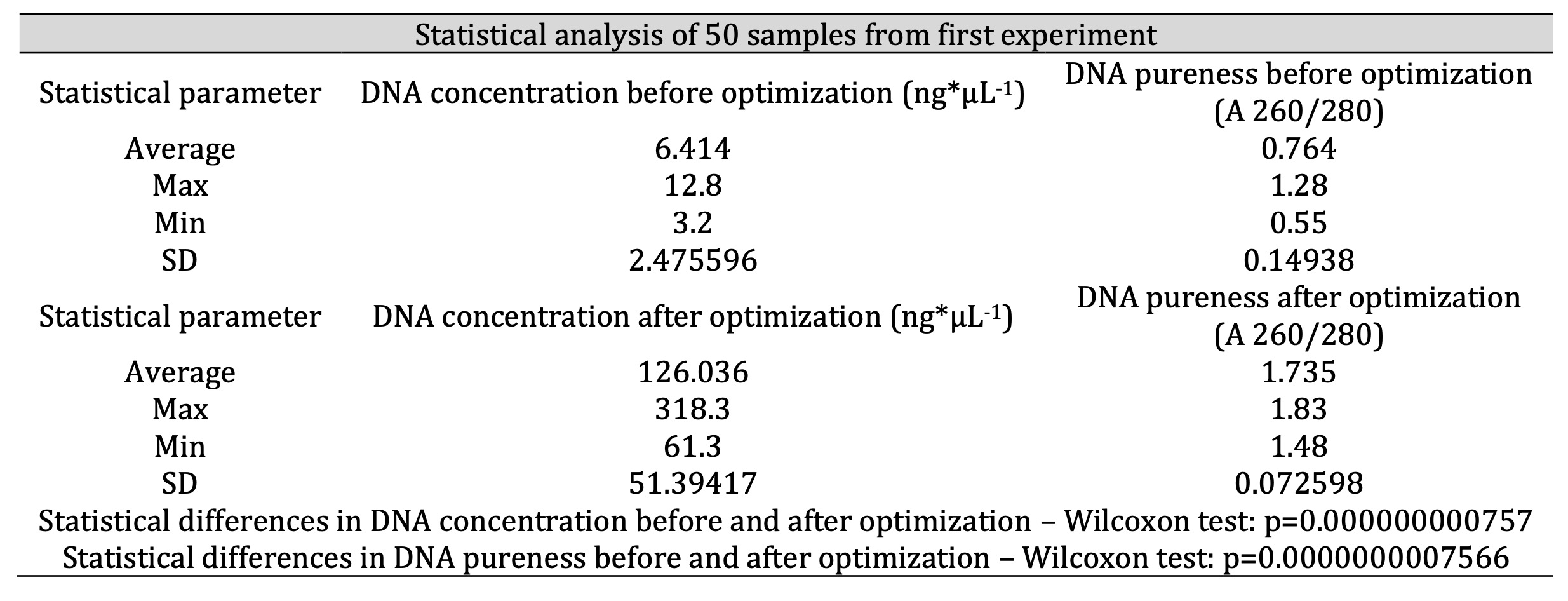
Table 1: Results of DNA isolation before and after optimization (statistics for 50 samples; n=50)
We experimented with two methods of drying the ethanol in the samples (step 10 above), 1) placing in an incubator for about 20 minutes at a temperature of 37°C and 2) standing in the open air for 20 minutes at room temperature [34]. Drying of ethanol in the open air at room temperature yielded stable values of purity in the range 1.6-1.9 (A 260/280). Drying in the incubator yielded more labile results (Fig. 1). However, comparing the results of DNA concentration and purity after drying, by using Wilcoxon statistical test indicated that method of drying of ethanol does not exert statistically significant influence on concentration and pureness of DNA isolated from blood samples (the lack of significant difference in the DNA concentration and pureness between two methods of drying; p=0.721 for concentration and 0.241 for pureness; Table 2, Fig. 1). The optimal range of purity for our experiments was established due to optimization and available data [35, 36].

Table 2: Comparison between two methods of drying of ethanol in context of DNA concentration and pureness (optimization on 50 samples; n=50)
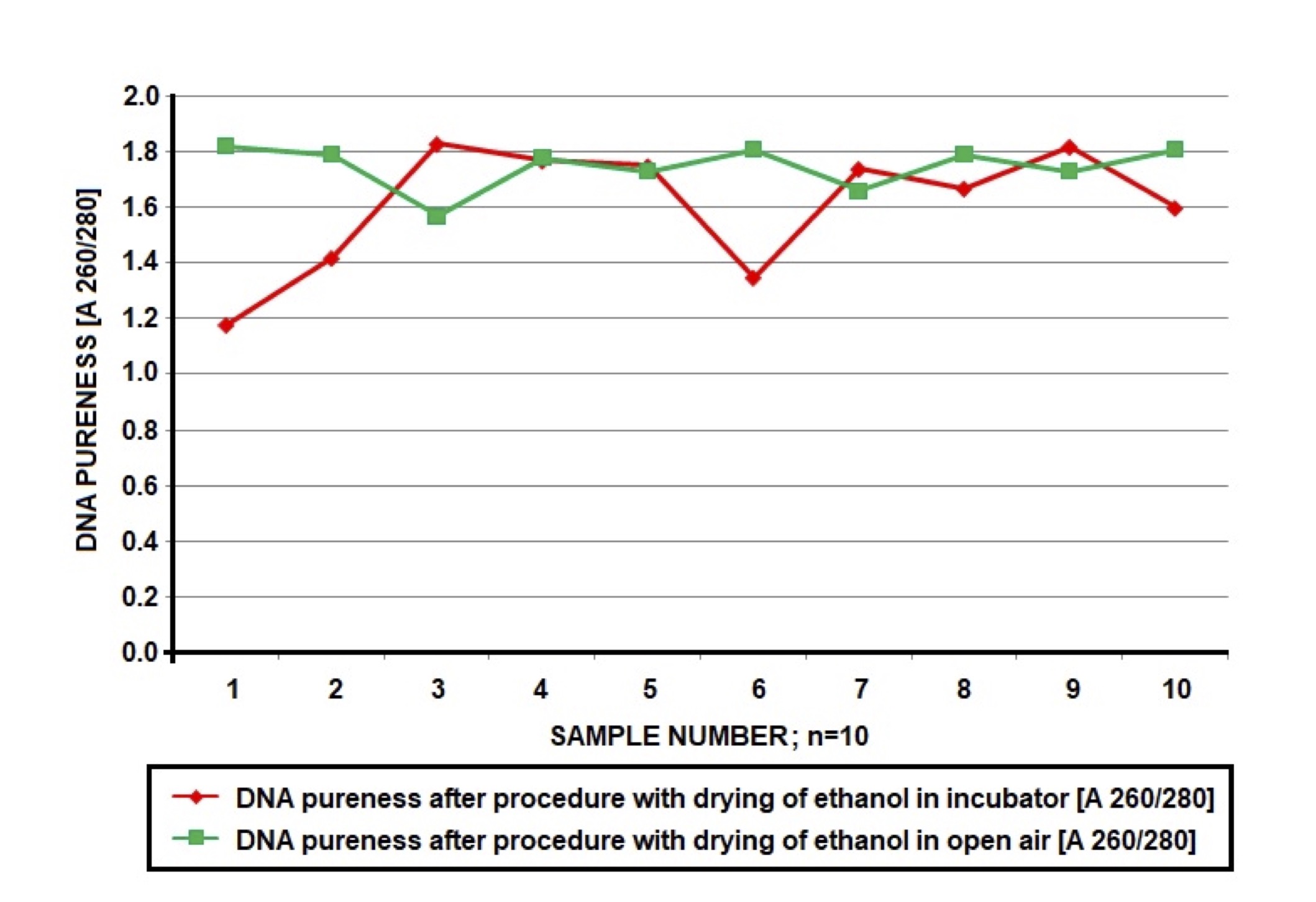
Fig. 1: Comparison of DNA pureness (A 260/280) of the same samples (total n=10x5=50) in which ethanol was drying in the incubator (20 minutes, 37oC, red marker) or in open air (20 minutes, room temperature, green marker). Conditions of drying influence on DNA pureness; incubation in open air produced more stable results in sufficient range of values 1.6-1.9 A 260/280.
After optimization we repeated the isolation protocol on the samples from the first experiment, achieving efficiency in range of 90-100%. Statistics for these results are presented in Table 1 (n=50). We established the optimal DNA concentration range (50-150 ng*μL-1) due to optimization and available data [37, 35, 31]. The purity of samples was in the range of 1.6-1.9 (A 260/280) in most cases. Figures 2 and 3 present graphical comparison of the DNA concentrations before and after optimization and DNA purity before and after optimization, respectively (the Wilcoxon test). There was a significant higher concentration (p=0.000000000757) and purity (p=0.0000000007566) of DNA after optimization which confirmed fundamental improvement achieved in DNA quality parameters in result of introduced modifications (Table 1, Figures 2, 3).
Samples with DNA concentrations above 150 ng*μL-1 were not problematic when diluted with TE Buffer. Depending on the DNA concentrations, we add TE Buffer 5 μL amounts, adjusting the concentrations to the optimum. The dilutions used are shown in the Fig. 4.
In our next experiment we analysed the efficiency of the optimized method in the isolation of DNA from two independent samples of whole blood: short-term frozen (n=50) and long-term frozen (n=50) (Table 3). All samples were frozen at the temperature of –80°C. We examined blood from vials with K3EDTA. These vials are used in every laboratory in the morphological tests. Our studies allow to use blood previously examined and does not require another blood sample for molecular tests [38]. Reducing the number of tubes and their volume (milliliters) of blood taken from the patient is important for patient safety especially for children and elderly [39, 40]. It is important to collect low volume of samples and obtain high efficiency method to isolate DNA, which allow scientists to perform multiple DNA tests on one sample, even from a long-term frozen blood [5, 31].
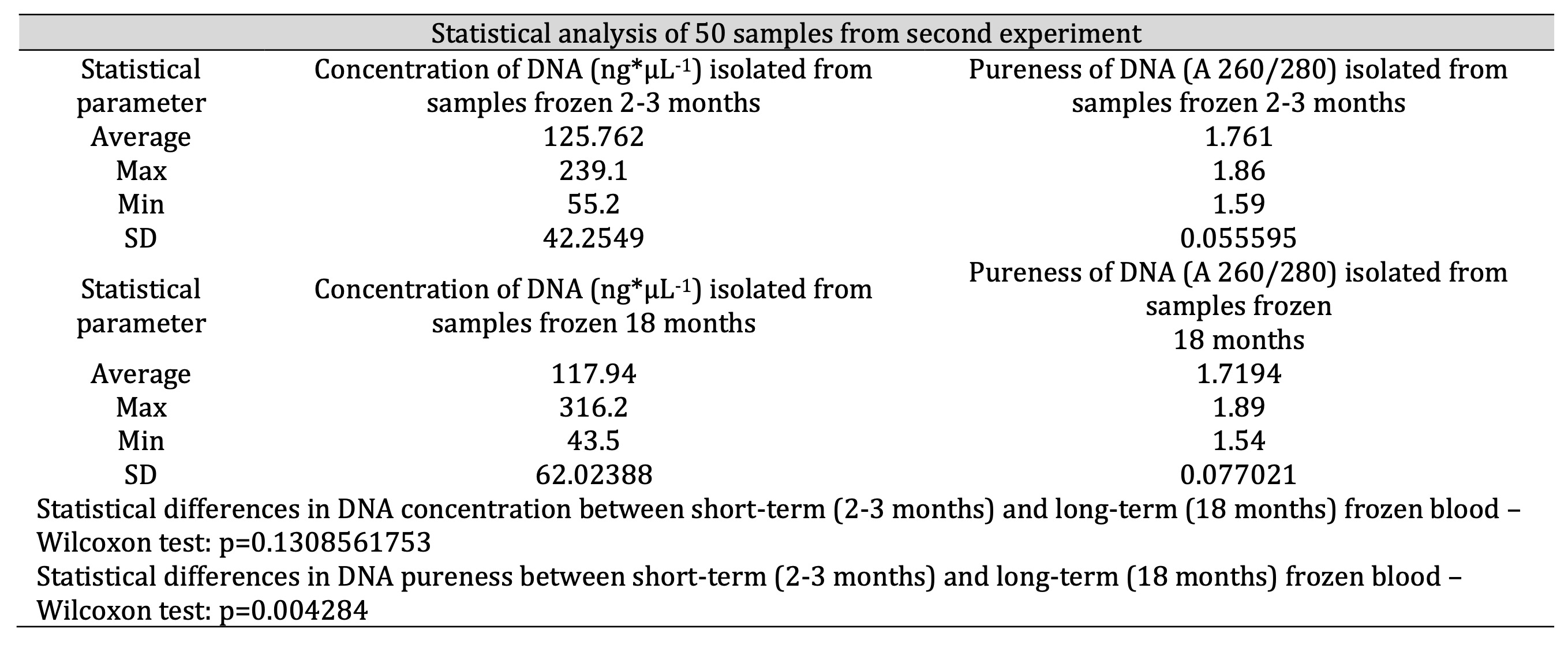
Table 3: Comparison of efficiency of DNA isolation – concentration (ng*μL-1) and pureness (A 260/280) – in case of two groups of samples: frozen 2-3 months and 18 months
The statistics (Table 3) confirm that the optimized method provided good efficiency of the DNA isolation both from short-term and long-term frozen whole blood. In the majority of samples we observed DNA the concentrations on the level of 125.762 ng*μL-1 for isolation from short-term frozen blood and 117.94 ng*μL-1 for isolation from long-term frozen blood (n=50). In general, the purity of the DNA also achieved satisfactory average values of 1.761 (A 260/280) for isolation from blood frozen for 2-3 months and 1.7194 (A 260/280) for isolation from blood frozen for 18 months (n=50). Blood frozen for 18 months yielded slightly lower quality of DNA resulting in a lower concentration and purity. We expected that prolonged storage may cause degradation of DNA and contamination of the isolation product. Nevertheless, Wilcoxon statistical test indicates that there is no significant difference in DNA concentration between short-term and long-term frozen samples (p=0.131). Therefore, in the matter of the concentration, optimized procedure enables isolation of DNA from both short-term and long-term frozen samples with comparable efficiency. However, Wilcoxon test showed that there is a significant difference in DNA pureness between samples frozen for 2-3 months and frozen for 18 months (p=0.004284). It suggests that long-term freeze actually worsen the pureness of DNA even when isolated with optimized procedure. Still most of analysed long-term frozen samples showed purity in range of 1.6-1.9 (A 260/280) and only few demonstrated purity slightly below mentioned range while purity and concentration of all 50 samples appeared sufficient for effective PCR-RFLP implementation (Table 3, Fig. 5). A graphic illustration of the efficiency of the optimized method in the matter of concentration and purity of DNA in these two sets of samples (short-term and long-term frozen) is presented in the Fig. 5.
The success of PCR amplification depends on the procedure of the DNA isolation. Fig. 6 shows electrophoresis of our isolation products, which provided suitable DNA for further research. The image on the left (Fig. 6) shows products achieved with primary isolation, when we used the original Epicentre protocol. The image on the right was produced from our optimized method. In the latter case we obtained un-degraded product, which we were able to use in further research. In our research we need to gain good quality and concentrated DNA, because we conduct PCR-RFLP. In lower concentrated DNA we had problems to gain a good material to gain visible products in the electrophoresis.
The results of spectrophotometric analysis of the isolated DNA confirmed that our optimized methodology was highly effective, with an efficiency at the level of 90-100% based in all of our experiments. The procedure yielded satisfactory concentration and purity of DNA. Furthermore, the efficiency was equally good when the whole blood had been frozen for 18 months. This was confirmed by electrophoresis of the extracted DNA samples in all cases (Fig. 6).
Discussion
In recent years nucleic acid isolation undergoes simplification and acceleration. In this context various systems may be characterized with certain advantages. E.g., the microfluidic platforms may be considered as cost-effective solutions for sample preparation in consequence of reduced sample and reagents size. Paper devices are generally inexpensive and easy to fabricate. Furthermore, such devices simplify the process of extraction by ensuring additional purification functions or by eliminate the need for centrifugation at certain steps. On the other hand, chitosan-based charge switching extraction techniques can be integrated to selectively bind target DNA or RNA from certain complex biofluids. It may result in rapid sample preparation at the detection area and increase extraction efficiency. However, a key task is to develop extraction strategies that may be applied to very diversified sample matrices. It may be important e.g. in case of newly emerging diseases when it is still uncertain which diagnostic medium is optimal [2].
The optimized method of DNA isolation allowed us to obtain DNA on the first attempt. Epicentre Master Pure™ DNA Purification Kit offers an easy and cheap method of DNA extraction from many materials (e.g. whole blood, tissues samples, fluid samples like saliva or semen, cell samples). The troubleshooting and optimization are time-consuming steps [4, 41]. The storage techniques of material are also fundamental [2, 4]. E.g., Zhou et al. (2022) [30] recommend the method of in situ cryo-silicification of whole blood cells for long-term preservation of DNA. These authors characterize their method as inexpensive and reliable. They also ensure that it yields cryo-silicified samples that meet the criteria for safe and long-term DNA preservation such as resistance to external stressors and stability even in unfavorable conditions. Finally, the researchers mention that their method facilitates highly effective freezing of DNA within thermally stable silica helping to avoid ultra-low temperature storage and accompanying cold chain inconveniences [30]. Our optimized method allowed us to obtain isolated DNA, which was ready for polymerase chain reaction (PCR) immediately. In our study we provided amendments to the standard protocol of DNA isolation. The instructions, which come with the kit do not contain sufficient detailed information (e.g. isopropanol temperature should be –20°C, incubation in the ice enhances the extraction of DNA, while RNA must be heated to room temperature). Dahm (2005) [7] previously suggested that a gentle pipette and vortex mixing should be used to dissolve the pellet, which we also include in our protocol (step 17).
Other changes resulted from multiple attempts such as increasing centrifuging speed, adding concentrated Tissue and Cell Lysis Solution, incubation in the freezer, drying and vortexing to dissolve isolated DNA. The results of our initial attempts and the final outcomes are shown in Table 1. The increase in DNA concentration and DNA purity were significant (Figures 2 and 3). We required DNA concentration in the range of 50-150 ng*μL-1 [37, 35], and DNA purity in the range of 1.6-1.9 ng*μL-1 (A 260/280) [36]. Our optimized protocol fulfilled both of these conditions.
Even the process of drying was problematic. In Table 1, it can be seen that the standard, supplied protocol resulted in a low amount of impure DNA (regardless the technique of drying). The results of optimization of this step are visible in Fig. 1 and Table 2. We tried to dry DNA in the incubator and in air both for about 20 minutes. Finally, both trials produced good results in matter of concentration and purity of DNA (no statistical differences between two methods of drying in Wilcoxon test that indicated p-values of 0.721 and 0.241 for concentration and pureness accordingly; Table 2), however the process appeared to be more effective stable for samples that were dried in air (Fig. 1). It is likely that drying in the incubator resulted in the contamination of DNA, due to biological or chemical factors [42]. Our laboratory have incubator which is used to multiple research, also to dry plants, heat probes etc. Probably forced air circulation in the incubator in small space allow to migrate pollutions, which could contaminate the samples. According to this we conclude, that in other laboratories could be similar problem. So we suggest, to dry DNA in the open air. We optimized the protocol on 50 samples, so we can conclude that the representativeness of our assessment can be considered as rather sufficient.
After optimization the concentration of DNA was high (Fig. 2); for some samples, it was even higher than 150 ng*μL-1. It is not possible to conduct PCR on highly concentrated samples [42]. Thus the samples were diluted according to the scheme shown in the Fig. 4. This procedure yielded a larger sample with an optimal DNA concentration. It was beneficial for optimizing conditions in PCR reactions, because we conducted many reaction from one sample. After one process of the isolation we could obtain more product for next analysis.
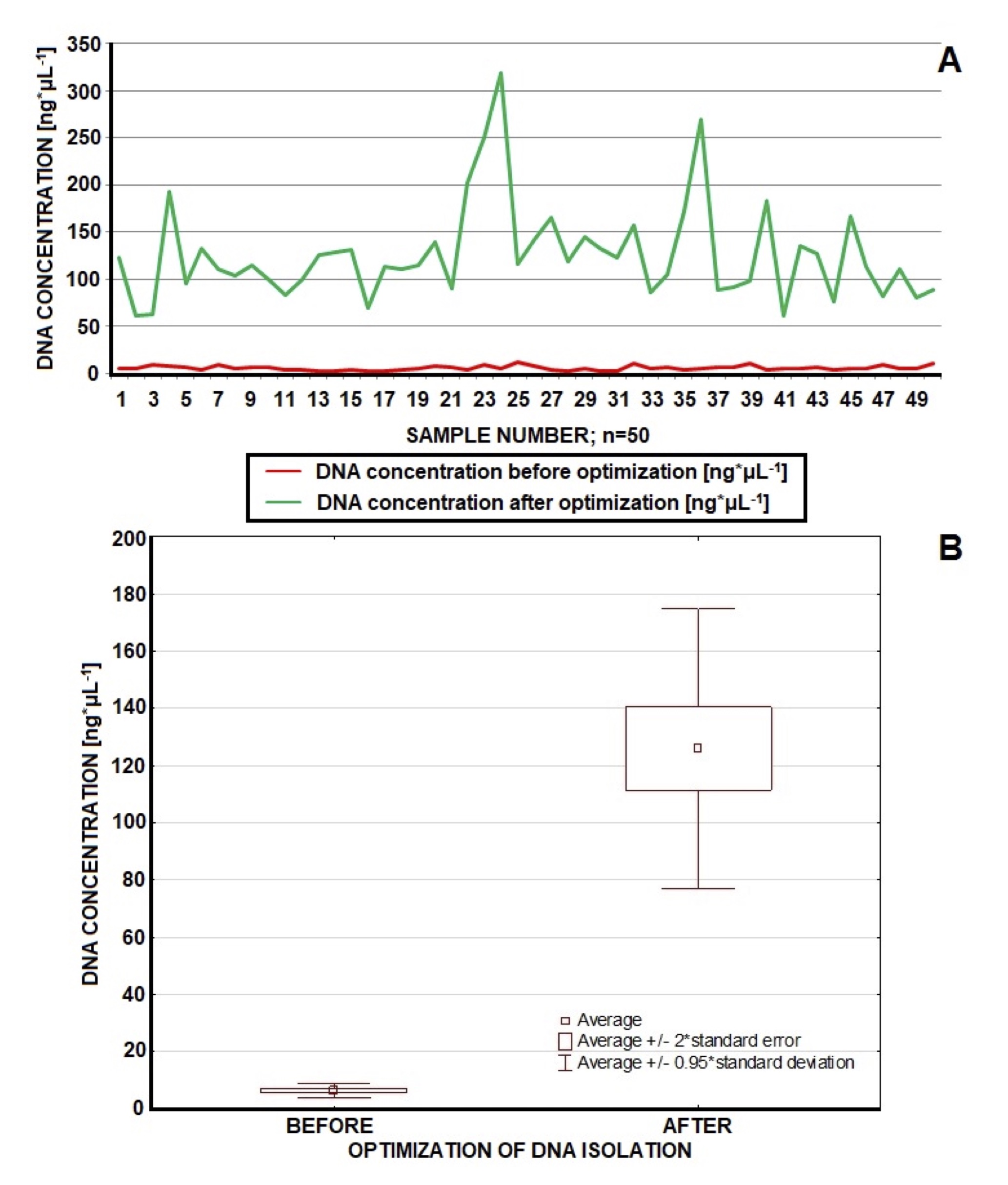
Fig. 2: Comparison of DNA concentration (ng*μL-1) before and after optimization (n=50). Before optimization we achieved quite stable but low values of DNA concentration (average of 6.414 ng*μL-1). After optimization DNA concentration was more diversified but mostly in the fine range of 50-150 ng*μL-1 (average 126.036 ng*μL-1); concentrations of few samples topped 150 ng*μL-1. Optimized procedure guarantees improvement in DNA concentration.
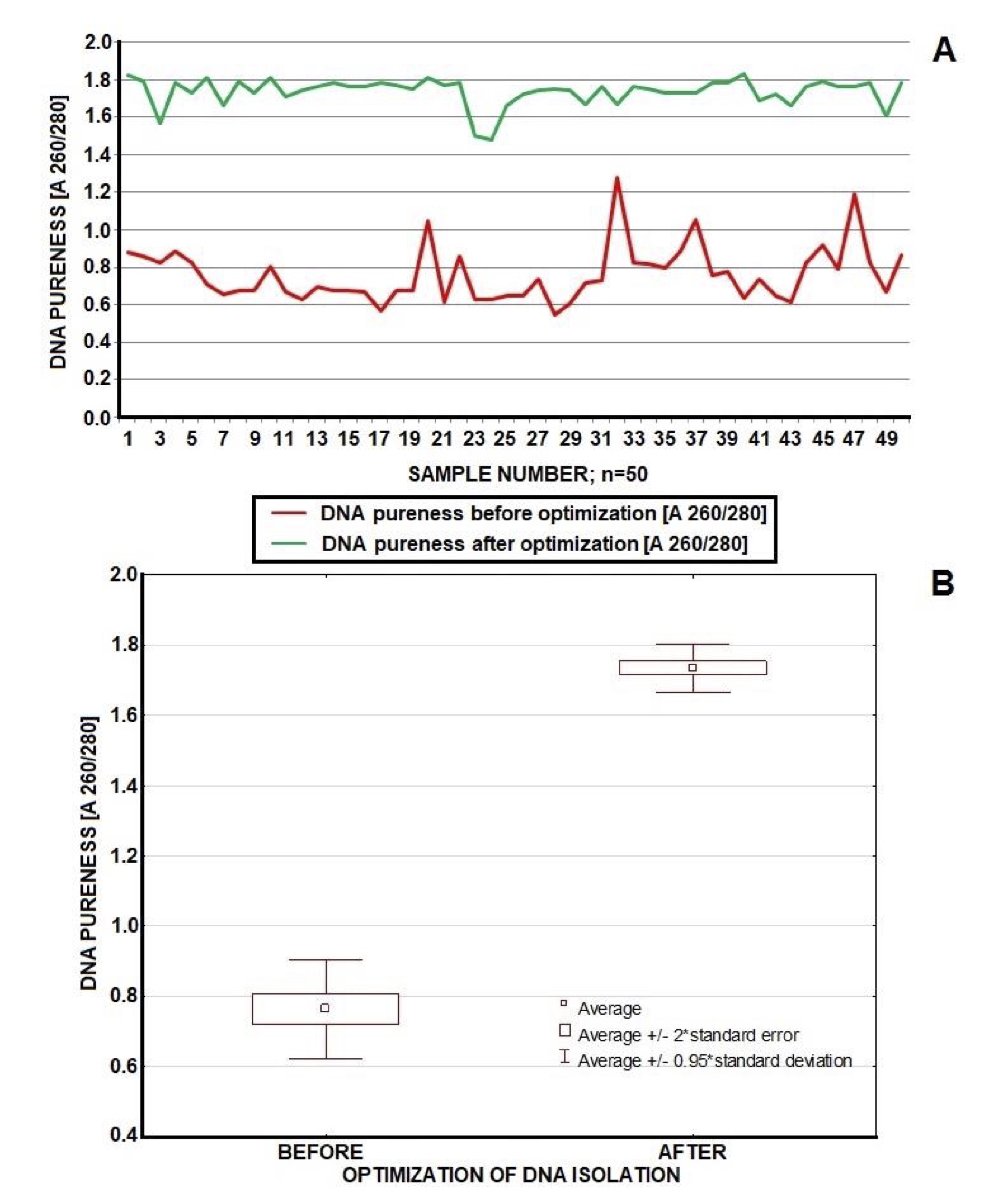
Fig. 3: Comparison of DNA pureness (A 260/280) before and after optimization (n=50). Optimized procedure guaranteed stable pureness mostly in a fine range of values (1.6-1.9 A 260/280). Contrary – un-optimal methodology produced unstable pureness below 1.5 (A 260/280). Optimization results in fundamental improvement of DNA pureness.
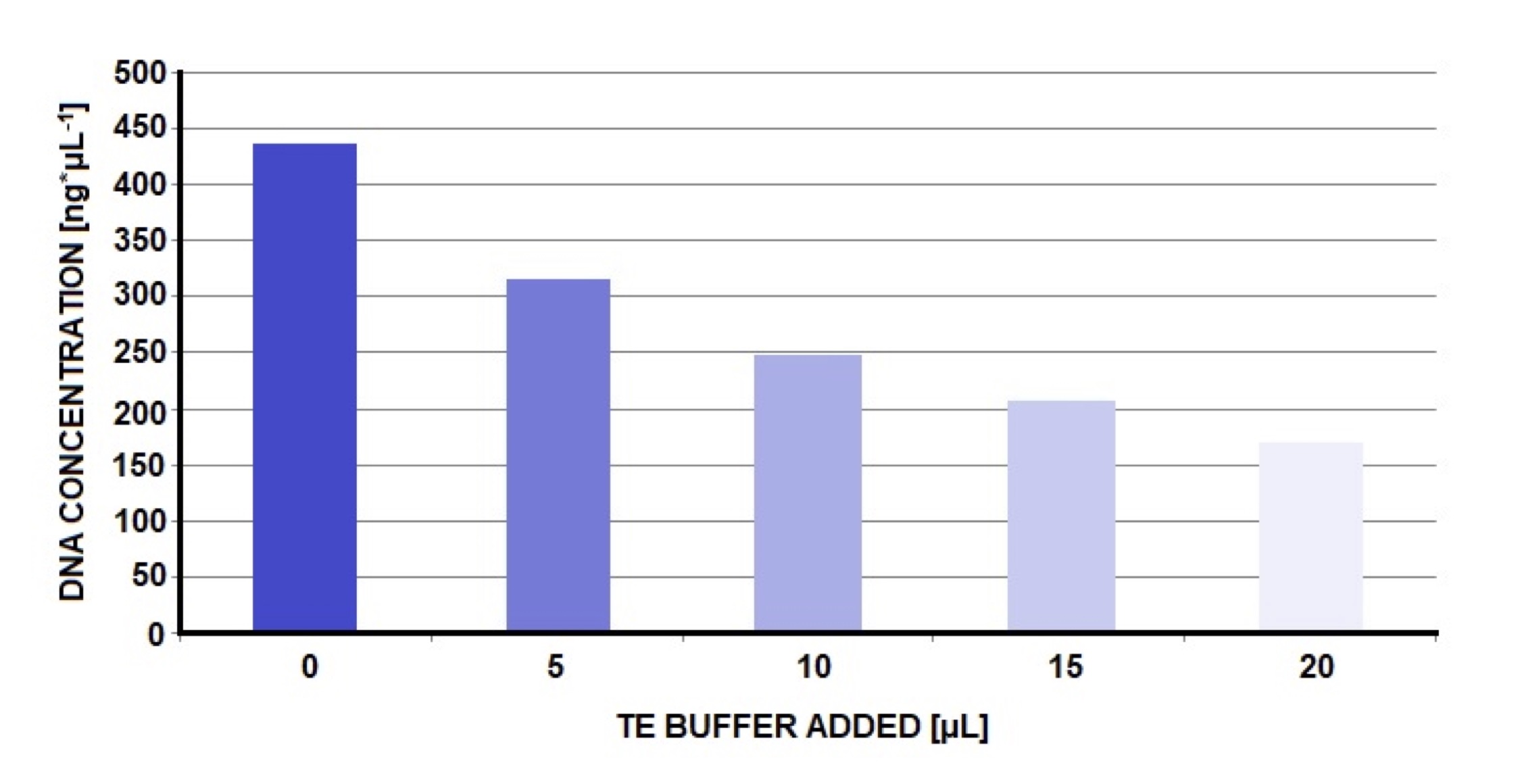
Fig. 4: Procedure of gradual dilution presented above was applied when achieved DNA concentration was too high. Dilution of DNA sample with TE Buffer enabled to obtain the value of concentration (ng*μL-1) close to optimum. In the beginning we observed relatively high DNA concentration (436.2 ng*μL-1). After adding 5 μL of TE Buffer DNA concentration fell to 317.3 ng*μL-1 (the next 5 μL resulted in 249.6 ng*μL-1). Finally, after adding 20 μL of TE Buffer (together in 4 steps) we obtained DNA concentration of 171 ng*μL-1 (the next dilution would certainly provide DNA concentration in the range of 50-150 ng*μL-1).
Our methodology was also tested on long-term stored blood. Freezing procedures generally tend to induce DNA strand breaks and DNA extraction yield may be reduced in consequence of freezing and thawing [4]. Table 3 shows the variation in DNA concentration and purity between short- and long-term frozen blood. Short- and long-term frozen blood samples yielded sufficient DNA to perform PCR but short-term frozen blood had a slightly higher concentration and purity on isolation. It has been shown that storage of whole blood in a freezer at a temperature of –80°C may not be as effective as storage in liquid nitrogen. DNA degradation proceeds faster at the higher temperatures in freezers [31, 43]. According to our results (as shown at Fig. 5), both short-term and long-term frozen blood samples could still yield sufficient DNA to perform PCR. However, the manufacturer notes that DNA isolation should be carried out from fresh material. Our methodology makes the kit also useful when using blood samples frozen for a longer period of time. Thanks to this, the Epicentre set has a wider range of applications, which is certainly a beneficial aspect for the manufacturer. Especially, that today specimens ale collected for a long time before they are examined. Often, multiple tests are performed from one sample. This is very important to have good method to isolation high-quality material from long-frozen time specimens [4, 31, 33, 34]. Today most methods concentrate on this aspect, because of its importance for the genomic research on the previously collected samples [44, 45]. Only a small number of the total samples yielded concentrations below established level of 50 ng*μL-1 and these were all derived from long-frozen blood (approximately 18 months), nevertheless they were still suitable for further analysis.
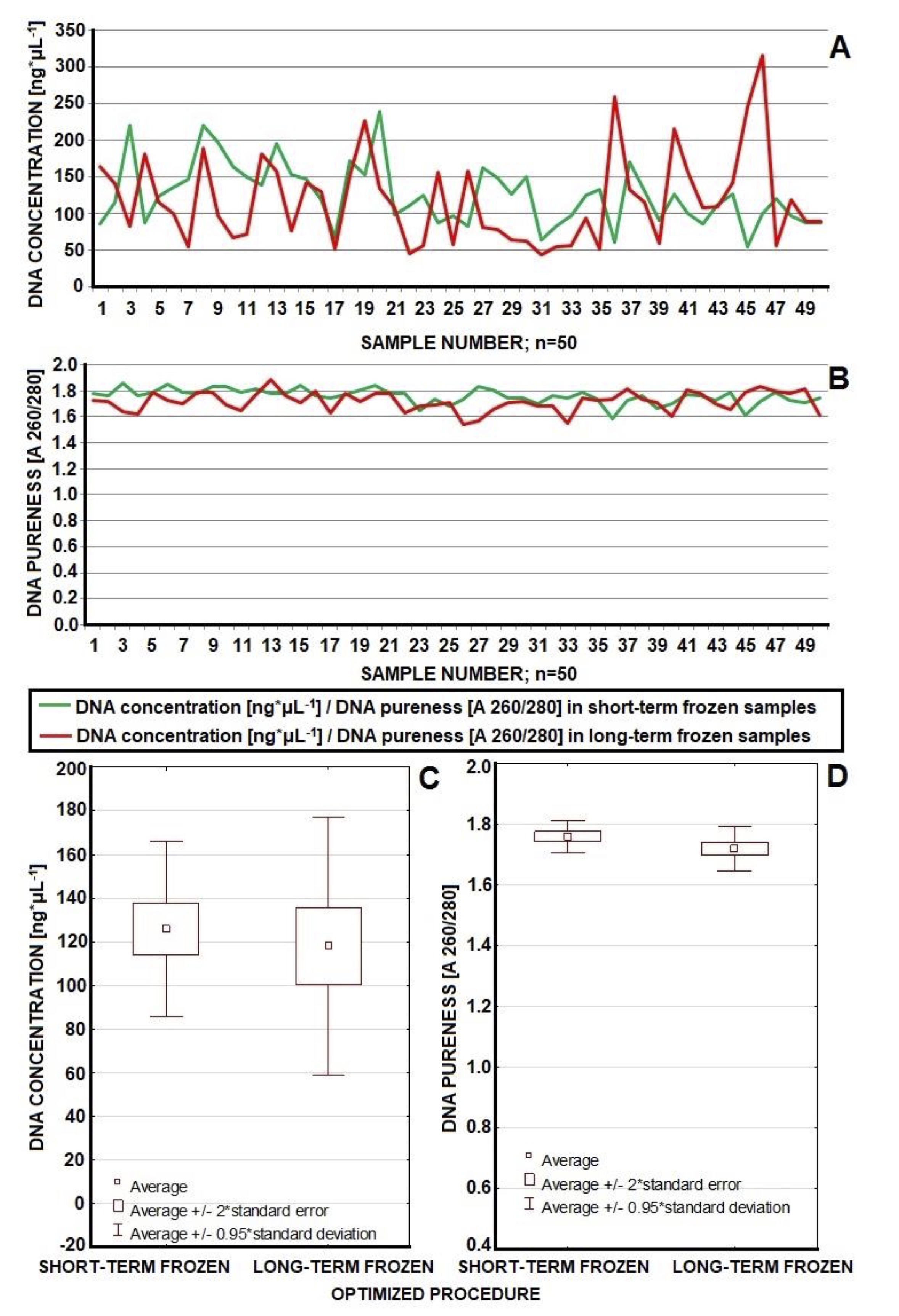
Fig. 5: Efficiency of optimized method in case of concentration of DNA isolated from short-term and long-term frozen whole blood (in both groups DNA concentration topped 50 ng*μL-1). Concentration in the majority of cases was placed in the range of 50-150 ng*μL-1 (A, C). Short-term frozen samples yielded better purity of isolated DNA while isolation from long-term frozen blood resulted in lower purity (statistically significant difference) (B, D). However all samples appeared suitable for further analysis. Optimization brought improvements in the concentration and pureness of DNA as well as enabled effective work with wider range of samples (both short-term and long-term frozen samples).
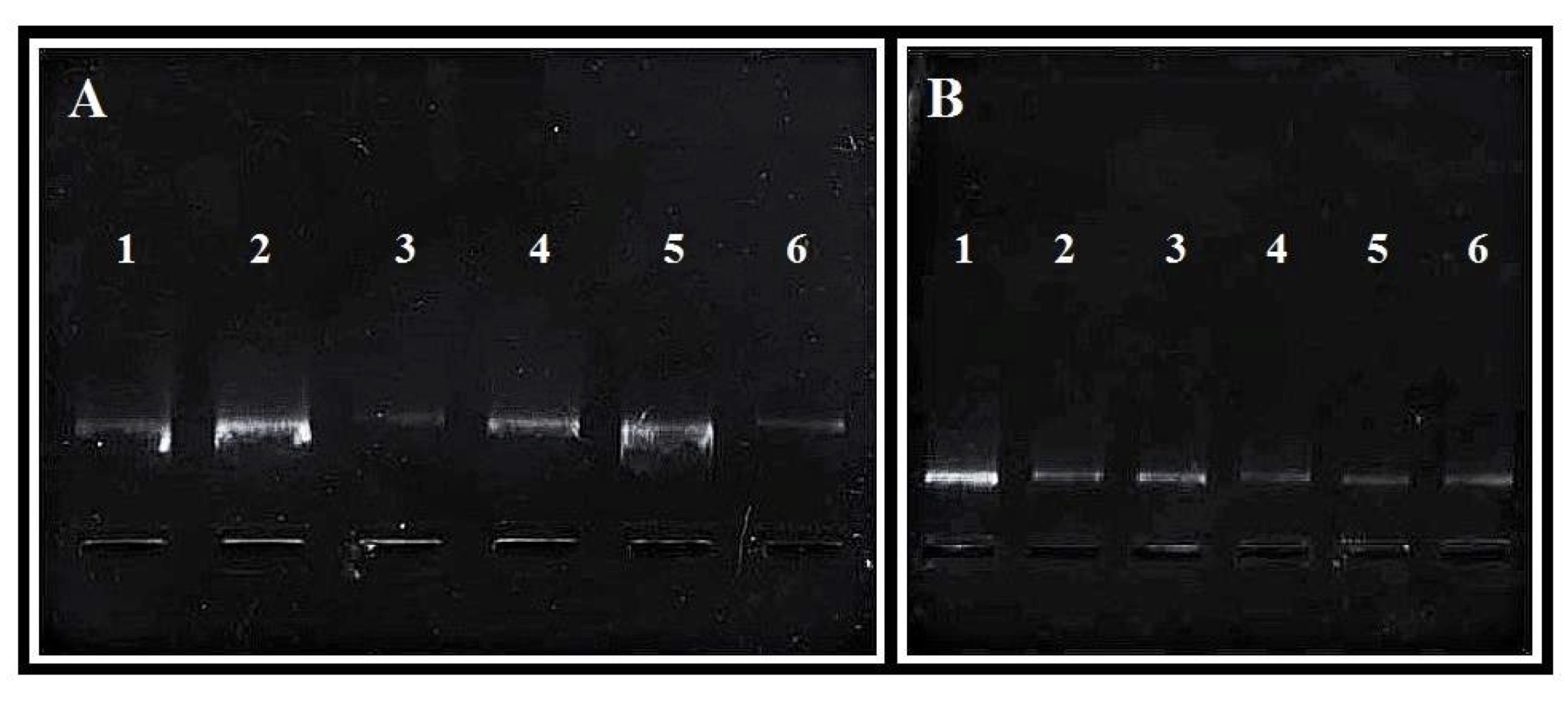
Fig. 6: Effects of electrophoresis of DNA isolation. On the left (A) obtained according to original Epicentre protocol (samples 1-6), on the right (B) obtained in optimized Protocol (samples 1-6). The image of undegraded product is a final confirmation of high effectiveness of optimized procedure.
Incorrect DNA extraction is a major limitation in PCR [46]. However, our optimized and original protocol did not use proteinase K. This enzyme is used to the lysis structural and membrane proteins [47, 48, 49]. It is useful in the isolate nucleic acids from various samples and obtain long fragments [47, 49]. K-proteinase is not recommended by the Epicentre in the procedure of DNA isolation from blood. We also resign with using this enzyme, because many publications show that the resignation did not affect the results [47, 50, 51]. Avoiding this part let us to save the time and shortens the duration of the procedure [47].
The presented results confirm that optimized DNA protocol fulfills the conditions of good extraction technique: it is relatively fast and easy to perform yet it guarantees a high repeatability, specificity and sensitivity. There are also no dangerous or harmful steps in the procedure (see [12]). Nowadays there is urgent need to adjust DNA isolation methods to requirements of point-of-care diagnostics which force to widely introduce the microanalytical systems (that could be used the nearest to patient as possible). Neither of currently use techniques may be considered as perfect for this task. One of fundamental steps of solid-phase extraction is centrifugation that requires specific machinery. On the other hand, extraction with magnetic beads needs external magnet source. Both solutions could be difficult to implement in point-of care systems. Additional difficulties would be connected for instance with maintenance of optimal temperature for critical procedure steps or with medical wastes utilization (see [11]). For that reason further improvement of DNA isolation techniques will be one of the most diligent matters in the near or possibly even distant future. Our results demonstrates the innovative and effective approach to the subject. It confirms a high effectiveness of method regardless of the duration of sample freezing, as well as the introduce important modifications (among others in matter of timing, temperature conditions, drying details) that make overall procedure more productive and relatively fast (absence of K-proteinase).
Epicentre Master Pure™ DNA Purification Kit for isolation, in accordance with the manufacturer’s recommendations, contains a safe and gentle protocol without hazardous chemicals. It also allows scaling the amount of reagents to obtain the appropriate sample volume and concentration of nucleic acid.
In conclusion, our optimized protocol for precipitation DNA isolation is one of the cheapest among those described. We checked the prices in the online stores. The isolation kit for processing 200 DNA samples and costs about $973 ($4.87 per sample), in solid-phase DNA extraction, isolation of 50 samples costs from $120 to $430 ($2.40-8.60 per sample). The cost of using the magnetic method is $620 for 96 samples ($6.45 per sample) or $645 for 48 samples ($13.44 per sample), but this method usually require specialized equipment (and do not allow for too much manipulation during the nucleic acid isolation process).
Taken together, all methods can be used for extraction DNA from blood and for other tissues. The time required for DNA isolation is approximately 30 minutes using the magnetic method, and from 20 minutes to 1 hour using columns (depending of the producer). DNA isolation using our method takes approximately 1 hour and 30 minutes.
New applications of optimization of the method of DNA isolation from human whole blood with Master Pure DNA Purification Kit™
Our optimized protocol differs from the standard Epicentre Master Pure™ DNA Purification Kit [32, 33] by including some additional specific manual steps:
Innovation 1. In step 4, we increased the spin time to 2 minutes and set the centrifuge speed at rcf=15295 x g. In step 6 we added 2x Tissue and Cell Lysis Solution to get more effective cell lysis. In step 9, we optimized the protocol by incubation in the freezer for 4 minutes.
Innovation 2. In the precipitation process we added pipetting in step 10, and we lengthened the vortexing from 10 seconds to 20 seconds. In step 13, we used chilled isopropanol (–20°C) rather than warm (see [40]). With cold isopropanol we got white strands of DNA faster. In the same step we added cooling on ice for 15 minutes.
Innovation 3. In step 16, we used cold ethanol (–20°C). Also, in the same step we removed the rest of ethanol by using a pipette and then left the tubes to dry for about 20 minutes. When all ethanol drops dried off we added TE buffer.
Innovation 4. In step 17, after suspension of DNA in 35 μL of TE Buffer we vortexed the sample tubes for 30 minutes. Then kept it overnight in room temperature before putting in a fridge (4°C).
Conclusion
- Optimized adaptations recommended in this paper allowed us to isolate DNA from whole frozen blood consistently, making any repeat of the protocol unnecessary.
- Our optimized method allowed us to obtain isolated DNA ready for PCR in a fast and effective way. Improvements in concentration and pureness of DNA were evident and significant (Wilcoxon test).
- We highlighted important deviations from the standard protocol recommended for DNA isolation (e.g. isopropanol temperature should be –20°C; incubation in ice, increasing centrifuging speed, adding concentrated Tissue and Cell Lysis Solution, incubation in freezer, drying and vortexing to dissolve isolated DNA).
- The process of dilution of DNA samples enabled us to obtain a greater sample volume with optimal DNA concentration. Establishment of specific scheme of solutions of DNA samples enabled us to obtain a greater sample volume with optimal DNA concentration.
- Short-term frozen (2-3 months) blood yields a higher DNA concentration of better purity than longer frozen material. In the matter of DNA concentration both short-term (2-3 months) frozen blood samples and long-term (18 months) frozen blood samples delivered satisfactory and comparable results of isolation (lack of significant difference in Wilcoxon test). In case of long-term frozen blood there was certain downgrade in DNA pureness when compared to short-term frozen samples (significant difference in Wilcoxon test). Nevertheless, DNA isolated from all tested samples was suitable for further analysis.
- All of short-term frozen blood Both short-term and long-term frozen samples yielded sufficient amount of extracted DNA to perform PCR.
-
The success of PCR amplification depends on the procedure of the DNA isolation protocol.
Electrophoresis of PCR products confirms that our optimized method yields sufficient DNA suitable for further research. - The results of spectrophotometric analysis of the isolated DNA confirmed that our optimized methodology was highly consistent (we estimated the efficiency at 90-100% based in all of our experiments). The procedure gives satisfactory DNA concentration and purity. In practice the efficiency was equally good when the whole blood was frozen for 18 months.
- To get final confirmation, we conducted electrophoresis on all DNA samples isolated in our experiments. We then conducted PCR on our samples. In all cases the PCR was successful.
Acknowledgements
We thank Professor Brendan P. Kavanagh (RCSI, Dublin, Ireland) for his help with improving English. We thank Prof. Maria Bogdzińska, and Karolina Hołderna-Bona, MSc, for their help in the laboratory analyses.
Author Contribution Statement
All authors (Sylwia Brodzka, Piotr Kamiński, Jędrzej Baszyński, Sławomir Mroczkowski, Katarzyna Rektor, Emilia Stanek, Joanna Kwiecińska-Piróg, Renata Grochowalska, Natalia Kurhaluk, Halina Tkaczenko) jointly participated in the experimental studies on the environmental relationships between nonenzymatic antioxidant defense and polymorphic changes in male infertility. They developed and participated in the development of the research problem and participated in the design of this article. All authors discussed the main theses of this work and revised the draft MS. They co-edited and revised the final version of the manuscript, conceived every part of this original article and participated in its design and coordination, and helped with every part of the MS project. Piotr Kamiński edited the entire text of the paper. All authors read and approved the final version of MS.
Declaration of Funding
This research received no funding. The authors declare no competing financial interests. The authors declare that this research did not receive any specific funding.
Statement of ethics and research authorization statement
All authors of this paper reporting experiments on humans confirm that all experiments were performed in accordance with relevant guidelines and regulations by Bioethical Commission of the Collegium Medicum of Nicolaus Copernicus University in Toruń, Poland (numbers of permissions as below). Subjects (patients) have given their written informed consent.
The study protocol has been approved by the research institute’s committee on human research.
All experimental protocols were approved by institutional and licensing committee, i.e. Bioethical Commission of the Collegium Medicum of Nicolaus Copernicus University in Toruń, Poland. In this paper informed consent was obtained from all participants of experiments, i.e. all samples were taken from people who gave written consent to use their blood for scientific research. The document „Consent to participate in research” was prepared according to international standard guidelines and approved by Bioethical Commission of the Collegium Medicum of Nicolaus Copernicus University in Toruń, Poland. This procedure is in line with international guidelines on compliance criteria.
The research was undertaken following to the Guidelines of the European Union Council and the current laws in Poland, according to the Bioethical Commission of the Collegium Medicum of Nicolaus Copernicus University in Toruń, Poland, under permit No KB 365/2015/May19/2015).
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Disclosure Statement
All authors of this manuscript have read the manuscript and have agreed to submit it in its current form for consideration for publication.
We confirm that neither the manuscript nor any parts of its content are currently under consideration or published in another journal. The authors, hereby assign to the journal, the copyright in the above identified article to be transferred, including supplemental tables, illustrations or other information submitted in all forms and media throughout the world, in all languages and format, effective when and if the article is accepted for publication. This paper has not been published elsewhere and it has not been simultaneously submitted for publication elsewhere in any language (this paper is not subject to any prior claim or agreement and is not under consideration for publication elsewhere). The article contains no libelous or other unlawful statements and does not contain any materials that violate proprietary right of any other person, company, organization, and nation.
The article was prepared jointly with other authors, and the authors agree with the authorship sequence. The participants in the study signed an informed consent form.
All authors of the manuscript have read and approved the MS of this paper and declare that it has not been published previously nor it is being considered by any other peer-reviewed journal. Participants gave written informed consent for publish this paper in its current form.
The authors have not any financial or non-financial or other potential conflict of interest and personal relationships with other people or organizations that could inappropriately influence (bias) this work. All authors have not any relationships or affiliations that may be perceived by readers to have influenced, or give the appearance of potentially influencing, what we wrote in submitted paper.
The authors declare that there is no any conflict of interest that could be perceived as prejudicing the impartiality of the research reported. All authors have contributed significantly, and are in agreement with the content of the manuscript.
References
| 1 | Srivastava S., Chatterjee B. Amplification of mitochondrial DNA fragment by PCR: Comparison of linear and circular DNA. Indian J Exp Biol 1995; 33: 153-154.
|
| 2 | Paul R., Ostermann E., Wei Q. Advances in point-of-care nucleic acid extraction technologies for rapid diagnosis of human and plant diseases. Biosens Bioelectron 2020; 169: 112592.
https://doi.org/10.1016/j.bios.2020.112592 |
| 3 | Alves Melo I.M., Pereira Viana M.R., Pupin B., Bhattacharjee T.T., de Azevedo Canevari R. PCR-RFLP and FTIR-based detection of high-risk human papilloma virus for cervical cancer screening and prevention. Biochem. Biophys Rep 2021; 26: 100993.
https://doi.org/10.1016/j.bbrep.2021.100993 |
| 4 | Yamagata H., Kobayashi A., Tsunedomi R., Seki T., Kobayashi M., Hagiwara K., Chen C., Uchida S., Okada G., Fuchikami M., Kamishikiryo T., Iga J., Numata S., Kinoshita M., Kato T.A., Hashimoto R., Nagano H., Okamoto Y., Ueno S., Ohmori T., Nakagawa S. Optimized protocol for the extraction of RNA and DNA from frozen whole blood sample stored in a single EDTA tube. Sci Rep 2021; 11: 17075.
https://doi.org/10.1038/s41598-021-96567-2 |
| 5 | Holzschuh A., Koepfli C. Tenfold difference in DNA recovery rate: systematic comparison of whole blood vs. dried blood spot sample collection for malaria molecular surveillance. Malar J 2022; 21, 1: 88.
https://doi.org/10.1186/s12936-022-04122-9 |
| 6 | Holmes F.L. Meselson, Stahl, and the Replication of DNA: A History of the Most Beautiful Experiment in Biology. New Haven: Yale Univ. Press, 2001; 1-503.
https://doi.org/10.12987/yale/9780300085402.001.0001 |
| 7 | Dahm R. Friedrich Miescher and the discovery of DNA. Developmental Biol 2005; 274-288.
https://doi.org/10.1016/j.ydbio.2004.11.028 |
| 8 | Meselson M., Stahl F.W. The replication of DNA in Escherichia coli. Proc Nat Acad Sci USA, 1958; 44: 671-682.
https://doi.org/10.1073/pnas.44.7.671 |
| 9 | Tan S.C., Yiap B.C. DNA, RNA, and protein extraction: the past and the present. J Biomed Biotechnol. 2009; ID 574398.
https://doi.org/10.1155/2009/574398 |
| 10 | Chacon-Cortes D., Griffiths L.R. Methods for extracting genomic DNA from whole blood samples: current perspectives. J Biorepository Sci Appl Med 2014; 2: 1-9.
https://doi.org/10.2147/BSAM.S46573 |
| 11 | Ali N., de Cássia Pontello Rampazzo R., Dias Tavares Costa A, Krieger M.A. Current Nucleic Acid Extraction Methods and Their Implications to Point-of-Care Diagnostics. Biomed Res Int 2017;9306564.
https://doi.org/10.1155/2017/9306564 |
| 12 | Shin J.H. Nucleic Acid Extraction and Enrichment. Adv Techn Diagn Microbiol 2018; Nov 10:273-292.
https://doi.org/10.1007/978-3-319-33900-9_13 |
| 13 | Maurya R., Kumar B., Sundar S. Evaluation of salt-out method for the isolation of DNA from whole blood: a pathological approach of DNA based diagnosis. Int J Life Sci Biotechnol Pharm Res 2013; 2: 53-57.
|
| 14 | Miller S.A., Dykes D.D., Polesky H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
https://doi.org/10.1093/nar/16.3.1215 |
| 15 | Fan M., Li Y., Chen J., Lin Y., Lai S., Peng S., Lin D., Wang J., Lu Y. Feng S. Plasmonic internal standard-decorated nitrocellulose membranes for duplex detection of circulating tumor biomarkers. Sens Actuators B Chem 2023; 395: 134508.
https://doi.org/10.1016/j.snb.2023.134508 |
| 16 | Toader G.A., Grigorean V.T., Ionita M. Solid Phase Oligo-DNA Extraction from Complex Medium Using an Aminated Graphene/Nitrocellulose Membrane Hybrid. Biomolecules 2024; 14, 3: 366.
https://doi.org/10.3390/biom14030366 |
| 17 | Katevatis C., Fan A., Klapperich C.M. Low concentration DNA extraction and recovery using a silica solid phase. PLoS One 2017; 12, 5: e0176848.
https://doi.org/10.1371/journal.pone.0176848 |
| 18 | Saiyed Z.M., Ramchand C.N. Extraction of Genomic DNA Using Magnetic Nanoparticles (Fe3O4) as a Solid-Phase Support. Am J Infectious Dis 2007; 4: 225-229.
https://doi.org/10.3844/ajidsp.2007.225.229 |
| 19 | Haddad Y., Xhaxhiu K., Kopel P., Hynek D., Zitka O., Adam V. The Isolation of DNA by Polycharged Magnetic Particles: An Analysis of the Interaction by Zeta Potential and Particle Size. Int J Mol Sci 2016; 17, 4: 550.
https://doi.org/10.3390/ijms17040550 |
| 20 | Haddad Y., Dostalova S., Kudr J., Zitka O., Heger Z., Adam V. DNA-magnetic Particle Binding Analysis by Dynamic and Electrophoretic Light Scattering. J Vis Exp 2017; 129: 56815.
https://doi.org/10.3791/56815-v |
| 21 | Bordelon H., Russ P.K., Wright D.W., Haselton F.R. A Magnetic Bead-Based Method for Concentrating DNA from Human Urine for Downstream Detection. PLoS One 2013; 8, 7: e68369.
https://doi.org/10.1371/journal.pone.0068369 |
| 22 | Berensmeier S. Magnetic particles for the separation and purification of nucleic acids. Appl Microbiol Biotechnol 2006; 73, 3: 495-504.
https://doi.org/10.1007/s00253-006-0675-0 |
| 23 | Gao X., Zhang K., Lu T., Zhao Y., Zhou H., Yu Y., Zellmer L., He Y., Huang H., Joshua Liao D. A reassessment of several erstwhile methods for isolating DNA fragments from agarose gels. 3 Biotech 2021; 11, 3: 138.
https://doi.org/10.1007/s13205-021-02691-1 |
| 24 | Ventura S.P.M., e Silva F.A., Quental M.V., Mondal D., Freire M.G., Coutinho J.A.P. Ionic-Liquid-Mediated Extraction and Separation Processes for Bioactive Compounds: Past, Present, and Future Trends. Chem Rev 2017; 117, 10: 6984-7052.
https://doi.org/10.1021/acs.chemrev.6b00550 |
| 25 | Flieger J., Flieger M. Ionic Liquids Toxicity-Benefits and Threats. Int J Mol Sci 2020; 21, 17: 6267.
https://doi.org/10.3390/ijms21176267 |
| 26 | Ghoshdastidar D., Senapati S. Dehydrated DNA in B-form: ionic liquids in rescue. Nucleic Acids Res 2018; 46, 9: 4344-4353.
https://doi.org/10.1093/nar/gky253 |
| 27 | Zhao H. DNA Stability in Ionic Liquids and Deep Eutectic Solvents. J Chem Technol Biotechnol 2015; 90, 1: 19-25.
https://doi.org/10.1002/jctb.4511 |
| 28 | Shabihkhani M., Lucey G.M., Wei B., Mareninov S., Lou J.J., Vinters H.V., Singer E.J., Cloughesy T.F., Yong W.H. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin Biochem 2014; 47, 4-5:258-266.
https://doi.org/10.1016/j.clinbiochem.2014.01.002 |
| 29 | Malsagova K., Kopylov A., Stepanov A., Butkova T., Sinitsyna A., Izotov A., Kaysheva A. Biobanks-A Platform for Scientific and Biomedical Research. Diagnostics (Basel) 2020; 10, 7:485.
https://doi.org/10.3390/diagnostics10070485 |
| 30 | Zhou L., Lei Q., Guo J., Gao Y., Shi J., Yu H., Yin W., Cao J., Xiao B., Andreo J., Ettlinger R., Jeffrey Brinker C., Wuttke S., Zhu W. Long-term whole blood DNA preservation by cost-efficient cryosilicification. Nat Commun 2022; 13, 1: 6265.
https://doi.org/10.1038/s41467-022-33759-y |
| 31 | Stanzick K.J., Simon J., Zimmermann M.E., Schachtner M., Peterhoff D., Niller H., Überla K., Wagner R., Heid I.M., Stark K.J. DNA extraction from clotted blood in genotyping quality. Biotechniques 2023; 74, 1: 23-29.
https://doi.org/10.2144/btn-2022-0061 |
| 32 | Epicentre® Technologies, Epicentre Protocols, Master Pure™ DNA Purification Kit. 2012: http://www.epibio.com/docs/default-source/protocols/masterpure-dna-purification-kit.pdf?sfvrsn=6
|
| 33 | Epicentre® Technologies, Epicentre Protocols, Master Pure™ DNA Purification Kit. 2012: http://www.epibio.com/docs/default-source/protocols/masterpure-dna-purification-kit-for-blood-version-ii.pdf?sfvrsn=8
|
| 34 | Oda Y., Sadakane K., Yoshikawa Y., Imanaka T., Takiguchi K., Hayashi M., Kenmotsu T., Yoshikawa K. Highly Concentrated Ethanol Solutions: Good Solvents for DNA as Revealed by Single-Molecule Observation. Chemphyschem: Eur J Chem Physics Physical Chem 2016; 17, 4: 471-473.
https://doi.org/10.1002/cphc.201500988 |
| 35 | Rajatileka S., Luyt K., El-Bokle M., Williams M., Kemp H., Molnár E., Váradi A. Isolation of human genomic DNA for genetic analysis from premature neonates: a comparison between newborn dried blood spots, whole blood and umbilical cord tissue. BMC Genet 2013; 14: 105.
https://doi.org/10.1186/1471-2156-14-105 |
| 36 | Ghantous A., Saffery R., Cros M.P., Ponsonby A.L., Hirschfeld S., Kasten C., Dwyer T., Herceg Z., Hernandez-Vargas H. Optimized DNA extraction from neonatal dried blood spots: application in methylome profiling. BMC Biotechnol 2014; 1: 14-60.
https://doi.org/10.1186/1472-6750-14-60 |
| 37 | Ahmad N.N., Cu-Unjieng A.B., Donoso L.A. Modification of standard proteinase K/phenol method for DNA isolation to improve yield and purity from frozen blood. J Med Genet 1995; 32: 129-130.
https://doi.org/10.1136/jmg.32.2.129 |
| 38 | Waters J., Dhere V., Benjamin A., Sekar A., Kumar A., Prahalad S., Okou D.T., Kugathasan S. A practical and novel method to extract genomic DNA from blood collection kits for plasma protein preservation. J Visualized Exp JoVE 2013; 75: e4241.
https://doi.org/10.3791/4241-v |
| 39 | Palmieri T.L. Children are not little adults: blood transfusion in children with burn injury. Burns &Trauma 2017; 5: 24.
https://doi.org/10.1186/s41038-017-0090-z |
| 40 | Hong N., Park J.Y. The Motoric Types of Delirium and Estimated Blood Loss during Perioperative Period in Orthopedic Elderly Patients. Biomed Res Int 2018; 9812041.
https://doi.org/10.1155/2018/9812041 |
| 41 | Guha P., Das A., Dutta S., Chaudhuri T.K. A rapid and efficient DNA extraction protocol from fresh and frozen human blood samples. J Clin Lab Analysis 2018; 32, 1: e22181.
https://doi.org/10.1002/jcla.22181 |
| 42 | Lorenz T.C. Polymerase Chain Reaction: Basic Protocol Plus Troubleshooting and Optimization Strategies. J Vis Exp 2012; 63: 3998.
https://doi.org/10.3791/3998-v |
| 43 | Lee S.B., Crouse C.A., Kline M.C. Optimizing storage and handling of DNA extracts. Forensic Sci Rev 2010; 22: 131-144.
|
| 44 | Goud T.S., Upadhyay R.C., Kumar A., Karri S., Choudhary R., Ashraf S., Singh S.V., Kumar O.S., Kiranmai C. Novel extraction of high-quality genomic DNA from frozen bovine blood samples by using detergent method. Open Vet J 2018; 8, 4: 415-422.
https://doi.org/10.4314/ovj.v8i4.11 |
| 45 | Mardan-Nik M., Saffar Soflaei S., Biabangard-Zak A., Asghari M., Saljoughian S., Tajbakhsh A., Meshkat Z., Ferns G.A., Pasdar A., Ghayour-Mobarhan M. A method for improving the efficiency of DNA extraction from clotted blood samples. J Clin Lab Analysis 2019; 33, 6: e22892.
https://doi.org/10.1002/jcla.22892 |
| 46 | Aygan A. Nucleic Acid Extraction from Clinical Specimens for PCR Applications. Turk J Biol 2006; 30: 107-120.
|
| 47 | Huijsmans C.J., Damen J., van der Linden J.C., Savelkoul P.H., Hermans M.H. Comparative analysis of four methods to extract DNA from paraffin-embedded tissues: effect on downstream molecular applications. BMC Res Notes 2010; 3: 239.
https://doi.org/10.1186/1756-0500-3-239 |
| 48 | Javadi A., Shamaei M., Mohammadi Ziazi L., Pourabdollah M., Dorudinia A., Seyedmehdi S.M., Karimi S. Qualification study of two genomic DNA extraction methods in different clinical samples. Tanaffos 2014; 13, 4: 41-47.
|
| 49 | Rezadoost M.H., Kordrostami M., Kumleh H.H. An efficient protocol for isolation of inhibitor-free nucleic acids even from recalcitrant plants. 3 Biotech 2016; 6, 1: 61.
https://doi.org/10.1007/s13205-016-0375-0 |
| 50 | Lever M.A., Torti A., Eickenbusch P., Michaud A.B., Šantl-Temkiv T., Jørgensen B.B. A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front Microbiol 2015; 6: 476.
https://doi.org/10.3389/fmicb.2015.00476 |
| 51 | Ghatak S., Muthukumaran R.B., Nachimuthu S.K. A Simple Method of Genomic DNA Extraction
|