Mechanisms of Senescence in Cancer: Positive and Negative Aspects of Cancer Cells Senescence
Keywords
Abstract
Introduction
Cellular senescence is a process during which cells lose their ability to divide. Senescent cells stop dividing but remain metabolically active and secrete many different substances into the environment. Characteristic markers of senescence are enlarged cell size and increased granularity, increased activity of the β-galactosidase enzyme (β-gal), increased levels of cyclin-dependent kinase inhibitors (p16, p21), increased levels of the p53 protein, inhibition of the cell cycle in phase G2/M, but also increased levels of critical epigenetic markers H4K20me3, H3K4me3, and a decrease in the level of H3K9me3, H3K27me3. In senescent cells, telomere shortening is also observed in successive rounds of replication, associated with reduced telomerase activity [1, 2]. Initially, cell senescence was considered an attribute of normal dividing cells. Recent studies suggest that cancer cells with unlimited dividing capacity may undergo a senescence process [3]. Researchers have shown that in cancer cells, senescence can be induced by inhibition of telomerase activity, induction of the DNA damage response [3], changes in the tumor microenvironment [4], regulation of senescence-related-proteins [3], irradiation [5], and changes in epigenetic modification and expression of epigenetic modulators [6]. The effect of cell senescence on tumor progression and invasiveness is ambiguous. Some studies suggested that induction of cancer senescence could inhibit cancer progression, but another indicated that induction of senescence could promote cancer progression and invasiveness. It is assumed that the role of cancer senescence in cancer progression and invasiveness is probably dependent on the molecular context and the specificity of some types of cancers [3]. Studies suggest that the ambiguous effect of senescence on cancer progression and invasiveness can result from mutations (e.g., PTEN) or protein status (e.g., p53) in some types of cancer [135].
Furthermore, components of the tumor microenvironment have been shown to influence senescence and its impact on tumor progression, metastasis and invasiveness [120-121]. It turns out that some therapies and compounds used in cancer treatment (chemotherapy, radiation therapy, CDK4/6 inhibitors, epigenetic modulators, immunotherapy, natural compounds, hormonal therapy) could induce cancer cell senescence. In the future, knowledge about molecular context of cancer cell senescence can play an essential role in the development of new more effective anticancer strategies and avoid the undesirable effects of the senescence process in cancer cells [7].
Senescence mechanisms in cancer cells
Telomerase activity inhibition
In contrast to normal cells, cancer cells have unlimited replicative potential or immortality. Tumor cell immortality may be related to activation of telomerase activity. Some studies have shown that hyperactivation of telomerase activity was observed in more than 85% of malignant tumors [8, 9]. The results indicated that genetic or pharmacological inhibition of telomerase activity in cancer cells could induce gradual shortening of telomeres and eventual cell senescence or apoptosis [10-13]. Sezmiya et al. (2002) showed that treatment of human monoblastoid leukemia U937 cells with a non-toxic dose of telomerase inhibitors (MST-312, MST-295, and MST-199) caused progressive telomere shortening and eventual reduction in growth rate and induction of β-galactosidase activity. However, a similar effect was observed after treatment of cellosaurus cell line cells with non-toxic doses of MST-312 [12]. In Barrett's adenocarcinoma SEG-1 cells, Shammas et al. (2005), after inhibition of telomerase activity, a marked reduction in median telomere length and complete loss of detectable telomeres in more than 50% of treated cells. Telomere loss caused senescence in 40% and apoptosis in 86% of treated cells [14]. Studies indicated that inhibition of telomerase activity and telomere shortening may lead to genome instability, a hallmark of cancer. Telomerase reverse transcriptase (tert) mutant zebrafish (tert -/-) have premature short telomeres and anticipate cancer incidence in younger ages. The zebrafish tert−/− mutant observed a high level of cyclin-dependent kinase inhibitors (cdkn1a, cdkn2a/b) and the inflammation marker TNF-α.However, in zebrafish, tert -/- embryos have shorter telomeres and display DNA damage responses that stabilize, p53 leading to premature ageing and death. Melanoma-derived cells in G2 tert −/− zebrafish embryos lead to increased tumor development. The melanoma developed in tert -/- recipients progress faster, reaching advanced stages faster and becoming more invasive [15] [Fig. 1].
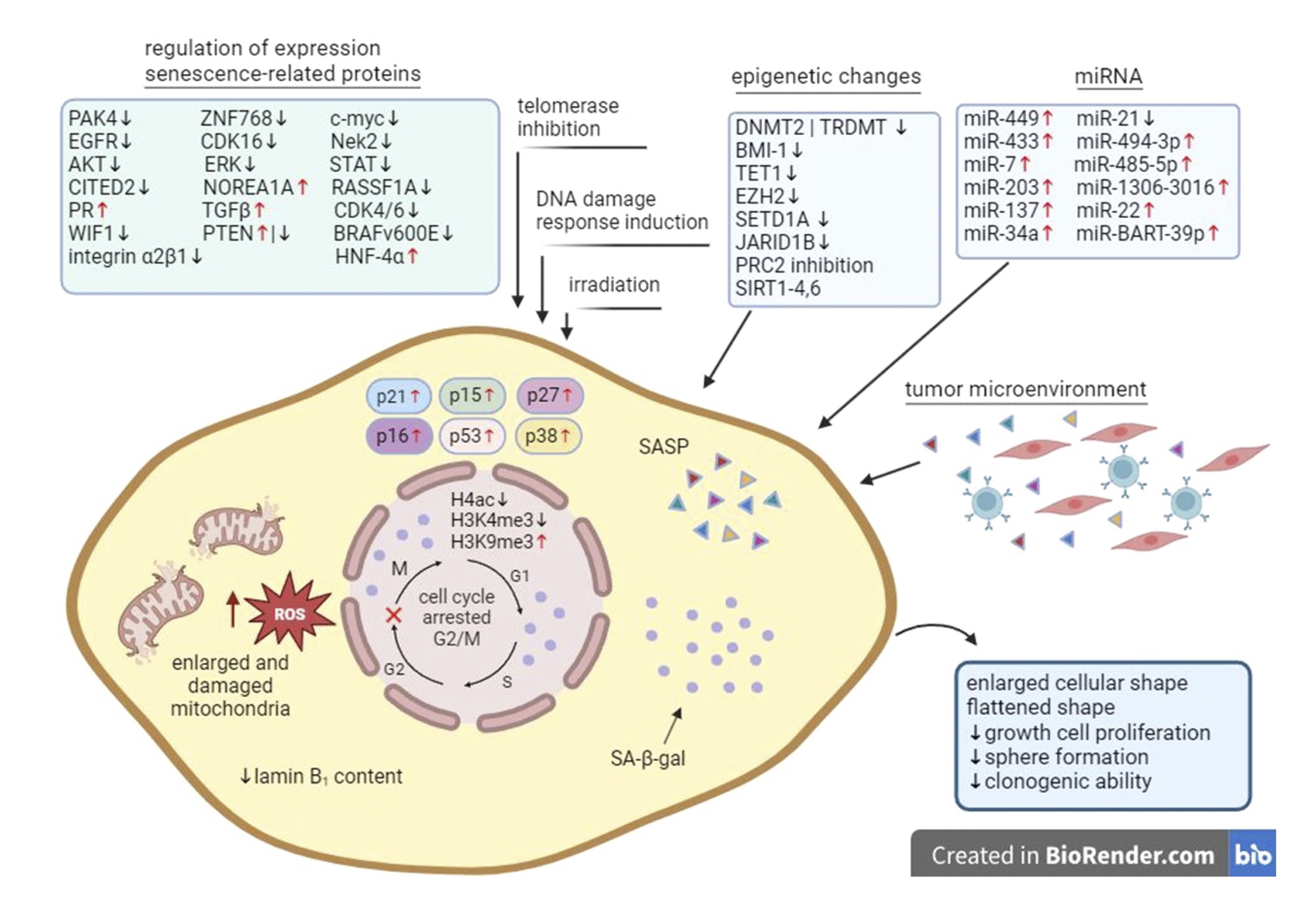
Fig. 1: Senescence mechanism in cancer cell. Cancer cell senescence could occur as a result of chemotherapy, radiation, inhibition of telomerase activity, induction of DNA damage, changes in the tumor microenvironment, regulation of senescence-related proteins, oxidative stress, inflammation, or epigenetic dysregulation. As a result of these factors, cell senescence occurs and can be observed through changes in cell morphology (enlarged cellular shape, flattened shape, enlarged and damage mitochondria), growth and cell proliferation, sphere formation, clonogenic ability, cell cycle arrested in G2/M phase, epigenetic modification (H4ac, H3K4me3, H3K9me3), cell cycle regulators (p21, p15, p27, p16, p53, p38) and changes in characteristic markers of senescence (laminin B1, SA-β-gal, SASP).
DNA damage response induction
The activation of DNA repair and cell cycle arrest is an immediate response to DNA damage, and these responses are designed to protect a damaged cell and promote its recovery. In the DNA damage response (DDR) mechanism, an important role is played by upstream DNA damage kinases (ATM, ATR), downstream protein kinases (CHK1 CHK2), DDR protein 53BP1, the mediator of DNA damage checkpoint protein 1 (MDC1), and NBS1 [16-19]. DNA damage can induce the expression of some proteins (p53, p16, p21), which prevent the growth of cells with severe DNA damage [20, 21]. Studies have shown that some compounds could induce DDR and, as a result, lead to cancer cell senescence. One of the DNA damaging agents is doxorubicin, which induces DDR through intercalation into DNA, DNA binding and alkylation, interference with DNA unwinding or DNA strand separation, DNA crosslinking, helicase activity and inhibition of topoisomerase II and generation of free radicals [22]. Recently, studies have shown that doxorubicin could induce DDR and promote cancer cell senescence. After doxorubicin treatment, Rouibah et al. (2021) showed induction of cell cycle arrest in the G2 / M cell cycle and senescence-associated secretory phenotype (SASP). The exit and senescence are regulated by both the INK4A / Rb pathways activated by p53-p21 and p38, probably due to their extensive crosstalk [23]. Hu et al. (2019) in human cervical cancer HeLa cells also observed an increased proportion of senescent cells, β-galactosidase activity, and SAPS after treatment with doxorubicin [24]. Induction of senescence after doxorubicin treatment was also observed in MCF-7 breast cancer cells [25] and hepatocellular carcinoma cells [26]. ROS have been shown to play a critical role in mediating genotoxic stress-induced DNA damage. Studies showed that resveratrol can induce premature senescence in lung cancer cells A549 and H460 through ROS-mediated DNA damage. In A549 and H460 cells, after resveratrol treatment, observed increased expression of senescence markers such as p53 and p21 protein, decreased expression of the EF1A protein, and positive β-galactosidase staining [27]. Similarly, icaritin could induce DNA damage by raising the ROS level and contribute to the senescence of HepG2 and Huh7 cells [28] [Fig. 1].
Tumor microenvironment
The tumor microenvironment (TME) is determined as non-cancerous cells present in the tumor. In the tumor microenvironment, including fibroblasts, adipocytes, immune cells, vascular and lymphatic endothelial cells, the extracellular matrix and proteins support the growth of cancer cells, which are produced by all cells in the tumor. In TME, cancer cells can frequently stimulate normal cells to produce some cytokines, chemokines, various growth factors, or matrix-degrading enzymes that consequently improve the malignancy of cancer cells [29]. Cancer-associated fibroblasts (CAFs) are the major component of the tumor microenvironment. Some studies have shown that CAFs are considered senescent cells and can contribute to the progression of various human cancers [4, 30-32]. Senescent fibroblasts, through cytokine secretion, can actively communicate with their microenvironment. This phenomenon was named the senescence-associated secretory phenotype. The cytokines secreted by senescent fibroblasts stimulate the growth, migration, and invasion ofpreneoplastic and malignant epithelial cells, but do not stimulate normal epithelial cells [33-35].
Yang et al. (2006) showed that the chemokine growth-regulated oncogene 1 (Gro-1) is a critical mediator of the stromal-epithelial interaction in tumor promotion through its induction of senescence in stromal fibroblasts. In NOF150 and NOF151 ovarian stromal fibroblast lines, Gro-1 treatment can lead to increased expression of the HP1 senescent marker HP1β in granular foci and levels of p16INK4A levels in the cytoplasm and increased senescence-associated β-galactosidase (SA-β-gal) activity. Furthermore, at least in part, Gro-induced senescence in cancer-associated fibroblasts can promote their tumor-promoting capacity [36].
Ren et al. (2013) observed that cancer-associated fibroblasts (CAF), but not normal fibroblasts (NF), secrete a high level of IL-6 with activated STAT3 and appear senescent in early passages in culture or cervical cancer tissues infected with high-risk HPV [31]. Some studies showed that oncogene-induced senescent cells through the SAPS phenotype can promote autocrine-like senescence in macrophages and other immune system cells. Senescent-associated macrophages can induce other immune system cells to evade tumor cell surveillance and senescent cell clearance. Furthermore, the accumulation of senescent cells promotes the release of SAPS, which contributes to tumor cell growth [37]. Marin et al. (2022) demonstrated that senescent cancer cells trigger strong antitumor protection mediated by antigen-presenting and CD8 T cells. This response is superior to the protection elicited by cells that undergo immunogenic cell death. Furthermore, patient-derived cancer cells exacerbate the activation of autologous tumor-reactive CD8 tumor-infiltrating lymphocytes (TIL) after the induction of senescence in these cells [38] [Fig. 1].
Studies showed that some combination of trametinib and palbociclib (T/P) could induce senescence in pancreatic ductal adenocarcinoma cells. Induction of senescence in cancer cells could stimulate tumor-reactive T cells, whose effect depended on SASP-mediated vascular remodeling. Treatment of T/P PDAC-associated CD8+ T cells expressed higher levels of the cell surface activation markers CD69 and CD44. Furthermore, CD8+ T cells isolated from the trametinib and palbociclib treatment cohort showed higher expression levels of the exhaustion markers PD-1, CTLA-4, 2B4, and LAG3. CD8+ T cells do not contribute to an antitumor response despite the initial influx of activated T cells into PDAC undergoing therapy-induced senescence [39] [Fig. 1].
Regulation expression of proteins involved in cell proliferation, migration, drug resistance, apoptosis, cell cycle and senescence induction
Recently, studies have shown that modifications in the expression of some proteins involved in regulating cell proliferation, migration, drug resistance, apoptosis, and cell cycle could induce cancer cell senescence. One of the proteins involved in these processes is the serine/threonine p21-activated kinase 4 (PAK4). The expression of this protein is frequently correlated with poor patient outcomes. Costa et al. (2019) in breast cancer cells, Hs578T and BT-549, have shown that the PAK4 protein could induce senescence through morphological changes, an increase in the SA-β-galactosidase activity, inhibit cell proliferation, and gene expression changes consistent with a SAPS phenotype. Furthermore, PAK4 could interact and phosphorylate RELB in serine 151, one of the NF-κB subunits. PAK4 interacting with RELB could inhibit RELB DNA binding, consequential transcription, and beta CCAAT-enhancer-binding protein beta (C/EBPβ). After inhibition of PAK4 in senescent breast cancer cells, in the most minor part, up-regulation of C/EBPβ [40, 41] [Table 1].
The Cbp / p300 interacting transactivator with carboxy-terminal domain 2 (CITED2) participates in the regulation of cellular cancer senescence. CITED plays an essential role in the regulation of other cellular processes such as proliferation, apoptosis, differentiation, migration, and autophagy [44]. Down-regulation of CITED2 in the lung cancer cell line A549 increased SA-β-gal activity and p21CIP1expression but also induced a senescence phenotype that included flattened and enlarged morphology [45] [Table 1].
Another group of proteins involved in the regulation of cell cycle progression and cancer cell senescence are cyclin-dependent kinases (CDKs). Dysregulation and mutation have been implicated in prostate, breast, cervical, and lung cancers. Jia et al. (2021) in lung cancer A549 and H1299 cells after CDK16 knockdown showed in these cell series senescence-associated phenotypes that included a decrease in cell proliferation rate, a blocked cell cycle in the G1 phase, and increased SA-β-gal activity [57]. Other kinases that play an important role in cancer cell senescence are CDK2 and CDK4/6. CDK2 siRNA and inhibition of CDK4/6 in MCF-7 resulted in inhibition of overexpressed phospho- C-MYC and hTERT and higher expression of SA-β-gal [58] [Table 1].
An essential role in the regulation of the cell cycle also plays NIMA-related kinase 2 (NEK2). Knockdown of NEK2 in a liver cell line, HepG2 cells, and the cellosaurus cell line, SMMC-7721 cells induced a senescent phenotype by decreasing the levels of cell proliferation and clone formation, increasing the percentage of cells in phase G0 / G1, decreasing the level of the phospho- AKT and phospho-Rb protein, and increasing the level of the p16 protein [59] [Table 1].
The Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR, and Jak/STAT pathways are often activated in human cancers. Recent research has suggested that inhibiting of these pathways could induce cancer cell senescence. In the breast cancer cell lines MDA-MB-468 and MDA-MB-231, the synergistic action of Akt and ERK through specific inhibitors induced cell senescence. Treatment of LY294002, U126, and NCTD breast cancer cell lines showed an increase in the percentage of cells in the G0/G1 cell cycle phase, an increase in the percentage of SA-β-gal-positive cells, and a lower expression of p21 expression [60]. Hayes et al. (2015) obtained similar results in PDAC tumor cells after treatment with ERK inhibitor, which also observed an increase in cells in G0/G1 and a decrease in cells in S and G2/M, reduced levels of cyclin D1, cyclin B1, p21 protein and hypophosphorylation and activation of RB [61]. Willobee et al. (2021) showed a similar effect after inhibition of MEK and CDK4/6 in the PDAC cell lines BxPC-3, Panc-1, and MiaPaCa-2, where a significant increase in phase arrest, inhibition of cell proliferation, and increase in β-galactosidase staining was observed [62]. In squamous cell carcinoma (KYSE30), combined MEK and STAT3 inhibitors induced cell senescence through cells arrested in phase G1, increased levels of SA-β-gal-positive cells [63] [Table 2].
Phosphatases play an important role in the regulation of kinase activity. PTEN phosphatase is the primary negative regulator of PI3K/AKT. Loss of PTEN induces cell senescence as a failsafe mechanism for defending against tumorigenesis, which is called PTEN-loss-induced cellular senescence (PICS). The loss of PTEN in MCF-7 caused an increase in AKT phosphorylation in serine 473 (S473) and threonine 308 (T308) and activation of mTOR1/2 complexes. These complexes phosphorylate the p53 protein in serine 15 (S15), increasing p21 expression. As a result, PTEN-depleted MCF-7 cells showed prematurely senescent phenotypes dependent on p53/p21 [64]. In the ovarian cell line, overexpression of PTEN promoted ageing by promoting p21 expression. Ke et al. (2021) showed that PTEN increased TRIM39 expression, which binds to p21 and inhibits p21 degradation [65]. In human lung adenocarcinoma A549 cells, siPTEN accelerated bleomycin-induced premature senescence by increasing the expression of the p16, p21 protein and increasing SA-β-gal activity. Furthermore, silencing of PTEN in A549 cells activated the PI3K/Akt/mTOR signaling pathway through increased phosphorylation of Akt, FoxO3a, mTOR, and accelerated cellular senescence [66] [Table 2].
Important roles in tumor initiation, growth, and metastasis play the (Wnt)/β-catenin pathway. Some studies indicated that Wnt inhibitory factor 1, one of the (Wnt)/β-catenin pathway components, could induce cell senescence. In salivary gland tumors, WIF-1 transfected cells (PA116, CaExPA79) observed a higher level of critical mediators of senescence (p21, p53, β-gal). WIF-1 could suppress salivary gland tumor cell stemming by reducing the number of stem cells, decreasing anchorage-independent growth, and decreasing the expression of master regulators of stem cell renewal, pluripotency (OCT4, c-MYC, WNT3A, TCF4, c-KIT, MYB) [68] [Table 2].
It was suggested that the depletion of the ZNF768 transcription factor could be related to the induction of cancer cell senescence.Studies showed that ZNF768 regulated cell senescence by modulating the expression of key cell cycle effectors and target genes. Loss of ZNF768 induced cell senescence in breast, cervical, and colon cancer, as evidenced by an enlarged, flattened shape and increased senescence-associated β-galactosidase activity. In glioma U87 cells, loss of ZNF768 induced high expression of genes that are part of the senescence-associated secretory phenotype (SASP) [42] [Table 3].
Studies showed that an essential role in cellular senescence may play the role of c-myc, a crucial regulator of various biological processes such as cell proliferation, growth, apoptosis, metabolism, adhesion, protein synthesis, DNA replication, and angiogenesis. In primary MYC-induced lymphoma and hepatocellular carcinoma, suppression of MYC induced SA-β-gal activity. In osteosarcoma cell lines, inactivation of MYC expression exhibited induction of p15INK4b, p21CIP mRNA, and the protein p16INK4a protein and a global reduction in total histone H4, reduction of trimethyl K4 histone H3 and increase of trimethyl K9 histone H3. Inactivation of MYC in hepatocellular carcinoma exhibited only induction of p15INK4b mRNA expression, resulting in a global reduction in total histone H4 acetylation. These changes suggested that c-myc plays an important role in osteosarcoma and hepatocellular carcinoma senescence [43] [Table 3].
Hepatocyte nuclear factor 4α (HNF4α, NR2A1) is a member of the nuclear receptor superfamily highly conserved. HNF4α is a crucial transcriptional regulator of genes, widely involved in xenobiotics, drug metabolism, and the development of gastrointestinal tract cancers. It was suggested that overexpression of this nuclear receptor in prostate cancer cells could induce cell cycle arrest and cellular senescence, resulting in their suppression of growth. The PC3-HNF4α-infectants (PC3-HNF4α-I) increased cell population in the G2/M phase without a significant presence of apoptotic cells as in the sub-G1 phase. However, significant SA-β-gal-positive cell increases were observed in PC-3-HNF4α and LNCaP-HNF4α infectants compared to their corresponding vector infectants. In HNF4α-I (AR-positive: LNCaP-HNF4α and VCaP-HNF4α; AR-negative: PC-3-HNF4α), mRNA and protein levels of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 (p21) increased markedly and frequently. In PC-3-HNF4α xenograft tumors, increased expression of the p21 protein was also observed. Furthermore, treatment with a senescence inducer in LNCaP and PC-3 cells could induce increased mRNA expressions of both HNF4α and p21. Knockdown of p21 activity in PC-3-HNF4α-I could significantly suppress the SA-β-gal and restore to the same levels as empty vector infectants. In HNF4α-I and shHNF4α-I (expresses wild-type p53) and HNF4α-I with HNF4 (expresses mutated p53), there were no significant changes in p53 protein levels, suggesting induction of cell senescence in prostate cancer cells independent of p53 [46] [Table 3].
Recent reports showed that some tumor suppressors or proto-oncogenes could influence cancer cell senescence. One of the tumor suppressors that plays a role in regulating cell senescence in cancer cells is NORE1A (RASSF5), a member of the RASSF family. Studies showed that lung tumor cell A549 contains an activated Ras gene and is wild-type for p53 but does not express NORE1A. After transfection of A549 cells, NOREA1A observed an increase in the activity of β-galactosidase and p21, suggesting that NOREA1A could be a potent inducer of senescence [47]. The tumor suppressor associated with cancer senescence that belongs to the RASSF family of tumor suppressors is RASSF1A. In RASSF1A-MCF-7 after doxycycline treatment, RASSF1A downregulates Erα induced SA-β-gal activity and increases the level of p21 Cip1 / Waf1 , characteristic of senescent cells [48] [Table 3].
The mutation of the BRAF-V600E proto-oncogene is a common occurrence in papillary thyroid cancer. However, BRAF-V600E expression can trigger oncogene-induced senescence (OIS) in normal and cancerous cells. This process causes BRAF-V600E cells to enter a cell cycle arrest state, which is achieved by using a negative feedback signaling mechanism. An increase in serum thyroid-stimulating hormone (TSH) due to hypothyroidism can cause senescent tumor cells to overcome OIS and proceed to malignancy. This highlights the importance of TSH/TSHR signaling in the development of papillary thyroid carcinomas (PTCs). An increase in TSH/TSHR signaling can lead to an increase in the levels of manganese superoxide dismutase (MnSOD) and dual-specific phosphatase 6 (DUSP6) enzymes, which in turn increases the production of oncogenic proteins (c-myc) [49]. Zou et al. (2015) showed that BRAFV600E induced senescence in BVE–PTC transplants by up-regulation of Trp53. Up-regulation of p53 was associated with solid activity of SA-β-gal, and the absence of cell proliferation due to the absence of Ki-67. This dependence has not been observed in primary BVE-PTC tumors [50] [Table 3].
CIP2A is an oncogene that inhibits protein phosphatase 2A (PP2A) and stabilizes c-Myc in cancer. Yang et al. (2018) suggested that CIP2A in hepatocellular carcinoma cells (HCC) promoted FOX1M dephosphorylation through PP2A phosphatase, which affects an increase in p21 and decreases CDK1, CDK2/4. These results suggested that inhibition of CIP2A inhibited cell cycle progression and promoted cell senescence by regulating transcriptional activity in HCC cells [51] [Table 3].
Recent studies have suggested that growth factors and their receptors may be essential in cancer cell senescence. Studies showed that progesterone receptors exhibit ligand-dependent antiproliferative effects by inducing cellular senescence in ovarian cancer. Treatment with PR-expressing progestin in ovarian cancer cells induced the expression of the p21 protein and transcription factor. Progesterone receptors with FOXO1 cooperatively upregulate p21 to promote cellular senescence by upregulating the expression of FOXO1 and proteins that regulate the cell cycle (p21, p15, p16, p27) and an increased level of β-galactosidase [52] [Table 3].
Some studies suggested that integrins could regulate cellular senescence. In human melanoma, SK/Mel/147 cells, the depletion of theintegrin α2β1 induces a senescence phenotype through the RAS / PI3K / Akt / mTORC1 / p53 / p21 pathway and decreases proliferation, invasive activity, and resistance to cell anoikis [67] [Table 3].
Xu et al. (2019) observed that administration reduced the level of epidermal growth factor receptor (EGFR) and could induce senescence, U87 glioblastoma cells, and osteosarcoma cells. Treatment with nutlin-3a of U87 and USOS cells influenced positive senescence-associated β-galactosidase staining, reduction of lamin B1, and reduced incorporation of 5-ethynyl-2′-deoxyuridine (EdU), p16. Furthermore, ectopic expression of EGFR in U87 or U2OS cells significantly attenuated senescence induced by p53 activation [53]. Interestingly, in OVCAR3 and A2780 ovarian cancer cells, EGFR could promote escape from carboplatin-induced senescence. Through SA-β-galactosidase staining in an ovarian cancer cell line, Li et al. showed that overexpression of EGFR inhibited the carboplatin-induced senescence-like phenotype. At the same time, EGFR knockdown may contribute to the carboplatin-induced senescence-like phenotype [54]. EGFR knockdown also induced senescence in gallbladder cancer cells GBC-SD and NOZ. siRNA-EGFR-induced cell cycle arrest in phase G0 / G1, increased SA-β-gal activity, up-regulation of p21 and p53 expression, reduction in the level of the phosphorylated form of Raf (c-Raf and b-Raf), MEK and ERK, and increased oxidative stress. Treatment with arctigenin, an EGFR inhibitor, induced the same effect as siRNA-EGFR in GBC cells and in xenografts in vivo [55] [Table 3].
Transforming growth factor-β (TGF-β) could be a potential senescence inducer in hepatocellular cell lines. TGF-β treatment of HepG2 and Hep3B cells could inhibit cell growth associated with flattened cell morphology and increase SA-β-galactosidase activity. In HCC cell lines TGF-β also causes the induction of p15Ink4b and p21Cip1 that is associated with c-myc downregulation, pRb underphosphorylation, p107 decrease and p130 [56] [Table 3].
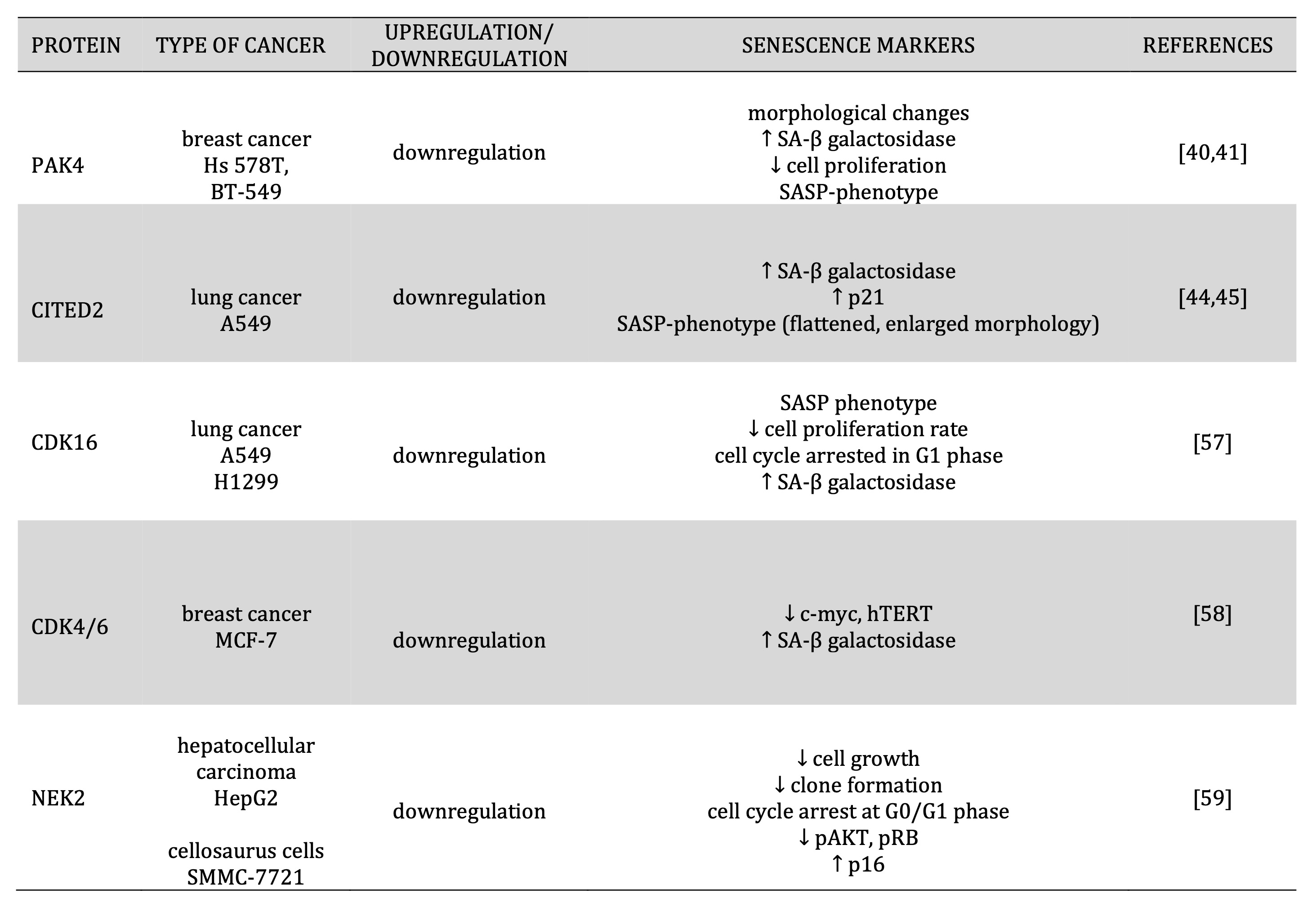
Table 1: Regulation expression of proteins involved in cell cycle regulation and senescence induction
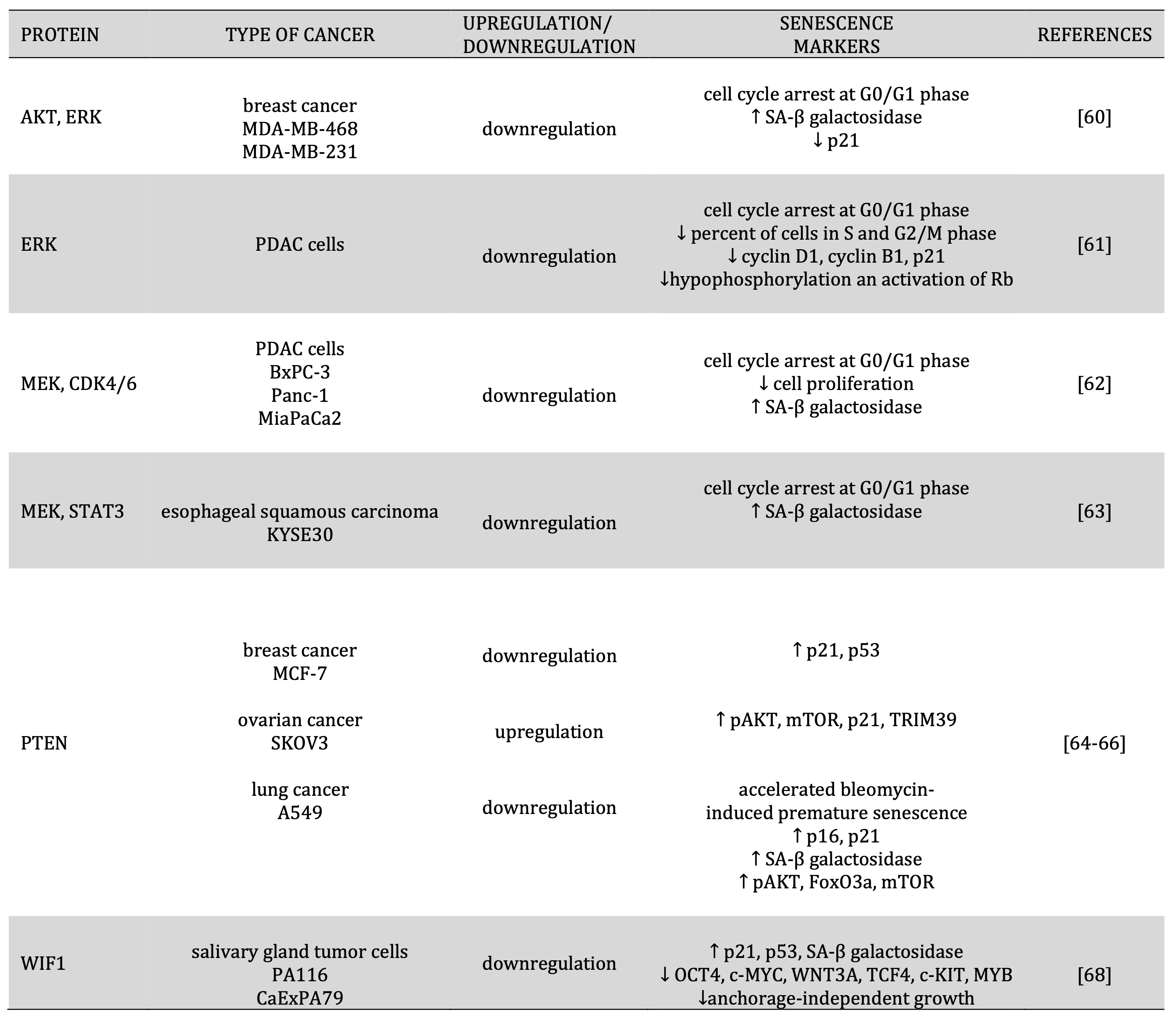
Table 2: Regulation expression of proteins involved in cell proliferation and senescence induction
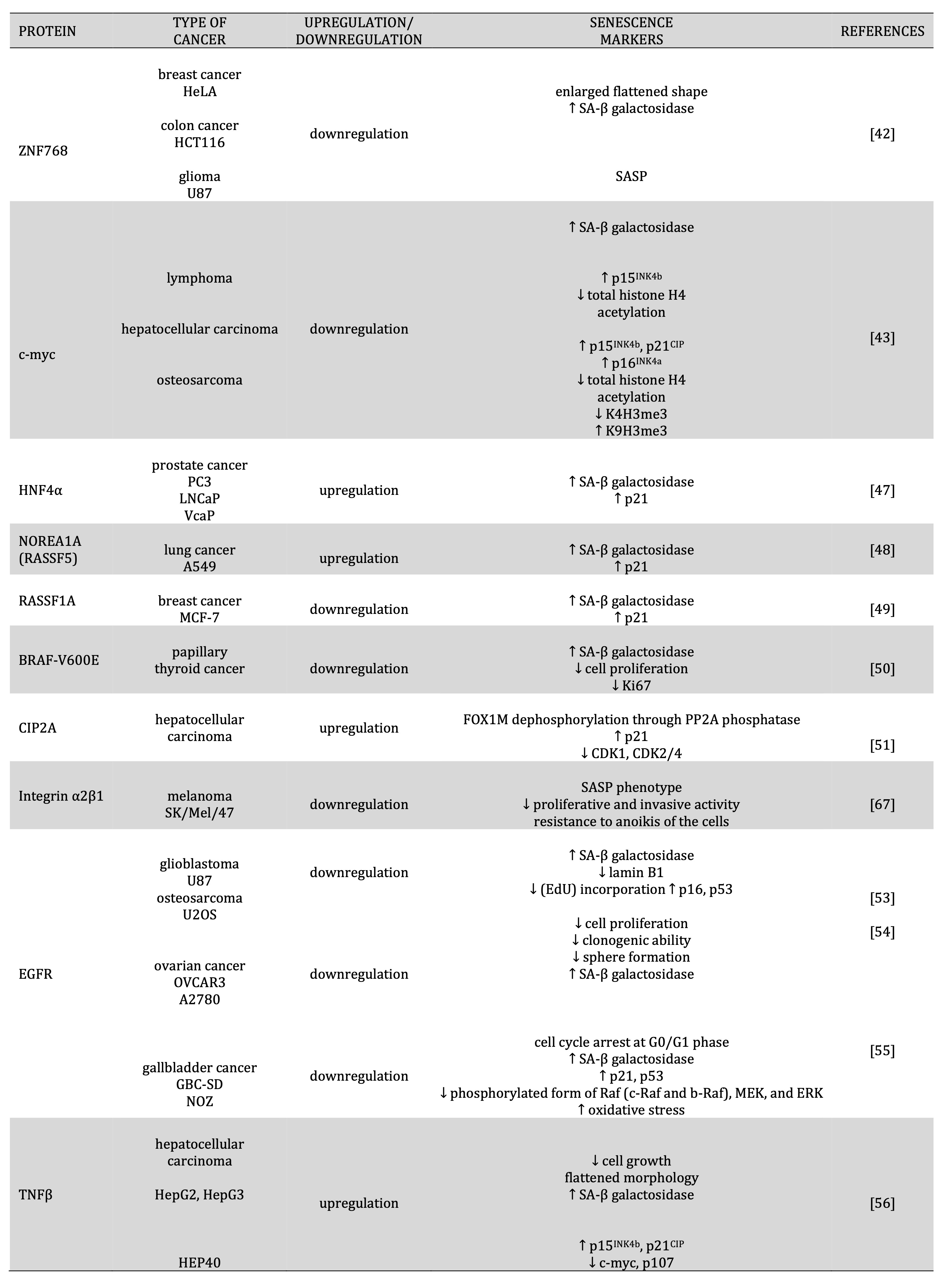
Table 3: Regulation expression of transcription factors, oncogenes, receptors, growth factors and senescence induction
Irradiation
Irradiation therapy is one of the approaches in cancer treatment. The central role of irradiation is to use high-energy radiation from X-rays, gamma rays, neutrons, protons, and nuclear energy to kill cancer cells and shrink tumors. Radiation therapy in cancer treatment induces DNA damage, inhibits tumor growth, and promotes necrosis and apoptosis [69]. Some tumor cells could escape cell death and undergo permanent cell cycle arrest-induced cancer cell senescence. Studies showed that irradiation could induce senescence in normal and cancer cells, but metabolic changes and secretory pathways are poorly known. Naginen et al. showed that after ionizing radiation from human colon cancer, HCT-116 cells exhibited typical senescence with enlarged cell size, intracellular granules, and positive staining for SA- β-gal, increased levels of p53 and p21. Furthermore, in senescent HCT-116 cells observed increased level of proinflammatory cytokines (IL-1α, IL-6, IL-8, IL-27, IL-15, IL-18), increased secretion of chemokines and chemo-attractants from inflammatory cells (CCL-5, CLXCL-10, CLXCL-1, CXCL-8, CCL-22, CCL-2), higher expression of extracellular matrix proteins (TIMP1/2/3; MMP-1), decrease in secretion of angiogenic agents VEGF-A and increase in the growth factors secretion (PLGF, HB-EGF, GM-CSF, M-CSF, PDGF-AA, PDGF-AA/AB, TGF-α/β1). Senescent HCT-116 cells observed changes in redox system metabolites, aerobic glycolysis, citric acid cycle metabolites, pentose phosphate pathway metabolites, amino acids levels, and polyamine metabolites. These observations showed that irradiation-induced senescence in cancer cells could influence metabolic and secretory pathways [5].
Essential roles in senescent induction play irradiation doses. In non-small cell lung cancer A549 cells, doses of 2 Gy were observed ~ 20% of SA-β-gal-positive cells, but 10 Gy doses generated ~ 80% of SA-β-gal-positive cells [70]. A similar dose of 10 Gy was sufficient to induce cancer senescence in wild-type p53 breast cancer MCF-7 cells [71]. Studies suggested that the status of p53 could be important forthe induction of senescence in cancer cells by irradiation. Cellular stress by ionizing radiation could stabilize the p53 protein, leading to the accumulation of this protein in the nucleus and the activation of some transcription factors. Translocation of the p53 protein to the nucleus in normal human cells causes a delay in cell cycle progression and activation of DNA repair. It could also induce premature senescence or cell death by apoptosis. Mirzayans et al. (2005) showed that in some types of cancer cell lines that express wild-type TP53, radiation can enhance nuclear TP53 levels, induction of transcription, and increase the CDKN1A protein level [72].
Furthermore, radiation exposure A172 (malignant glioma), HCT116 (colon carcinoma), SKNSH (neuroblastoma), and wild-type 53 cancer cells resulted in accelerated senescence. Accelerated senescence in these cell lines evaluated by β-gal expression and DNA, delayed cell cycle progression, acquisition of flattened and enlarged morphology, and sustained growth inhibition [72]. In MDA-MB-231 breast cancer cells, attenuated p53 irradiation did not induce senescence but cell death by apoptosis [71].
Another factor that could be important in the induction of cancer cell senescence by irritation is the status of securin. Seucurin is a protein involved in DNA replication, repair, and tumorigenesis. It has been found that irradiation of wild-type colon cancer cells induced apoptosis but induced senescence and enhanced radiosensitivity in securin-null cells. Radiation in securin-null HCT116 cells increased the level of g-H2AX, phospho-Chk2 (Thr-68), p53, p21, and decreased the level of Rb [73].
Tumor radioresistance is one of the problems in cancer treatment because it leads to cancer recurrence, metastasis, and poor survival in cancer patients. CDC6 has been identified as a potential prognostic biomarker of radioresistance. In radioresistant CNE2-R cells, acute exposure to IR elevated CDC6 levels by preventing the ubiquitin-proteasome degradation pathway protein. CDC6 depletion enhanced IR-induced senescence, showing typical senescence morphology such as cell size, cell enlargement, positive β-galactosidase staining, and elevated p16 and p21 levels. An in vivo study validated that CDC6 depletion resensitized radioresistant cell xenografts to IR treatment [74].
Schoetz et al. (2021) showed induction of senescence in the panel of HNSCC cell lines after radiation at 0–8 Gy for 8 days. Most radioresistant cell lines (UPCISCC 040, Cal 27, and Cal 33 cells) showed an early and strong induction of senescence (2 days after irradiation). Then, they declined by the disintegration of senescent cells and the repopulation of viable cells. More sensitive radiation cell lines (UDSCC 2, UPCISCC 099, UPCISCC 154, UPCISCC 131) exhibited a weaker senescence response. However, it was shown that senescent cells could support the proliferation of neighboring cells via released SASP factors in the tumor microenvironment. The conditioned medium of irradiated, senescent UPCISCC 040 cells significantly increased the clonogenic survival of Cal 27 cells compared to the conditioned medium of viable, non-senescent UPCISCC 040 cells [75] [Fig. 1].
Epigenetic changes
Some studies have suggested that epigenetic changes in senescent cells were crucial for the induction, progression, and maintenance of senescent cells. Recent data showed that epigenetic modification could also induce cancer cell senescence, but its role is not fully understood.
One of the epigenetic modifications is DNA methylation, which promotes transcriptional silencing. The enzyme responsible for DNA methylation is DNA methyltransferase (DNMT). Its decreased expression was observed in some senescent cells. DNMT2/TRDMT1 is a DNA methyltransferase that can potentiate its role in cancer cell senescence and be associated with chemotherapy-induced senescence. In glioblastoma cells with DNMT2/TRDMT1 knockdown, doxorubicin and etoposide mediated senescence with decreased SA-β-gal-positive cells and nuclear p21 pools [6]. Recent studies indicate that a class of methylcytosine dioxygenases, termed ten-eleven translocation (TET) family proteins, can erase existing DNA methylation. One of the epigenetic changes is the aberrant activity of the Polycomb Repressive Complex 2 (PRC 2) and the deregulated expression of the target genes. In the PRC2 complex, an important role in gene silencing through H3K27me3 plays an EZH2 protein. In human melanoma UCD-Mel-N, SK-Mel-173, and SK-Mel-119 cells, EZH2 silencing induced senescence features (flattened and enlarged morphology, SA-β-gal-positive cells, SAHFs, H3K9me3, increased cells in G1 phase and p21 upregulation) [76]. In triple-negative breast cancer (TNBC), EZH2 decreased TET1 expression by regulation of H3K27me3.Experimentally, targeting EZH2 in TNBC could induce cancer cell senescence by cell cycle arrest, increasing TET1 expression and activation of the p53 pathway. In the TNBC cells, after using a small-molecule inhibitor with EZH2 specificity, GSK343 cells observed changes reminiscent of cellular senescence, such as enlarged and flattened cell shape SA-β-gal positive cells [77]. Filipczak et al. (2019) observed a similar effect in lung cancer cells (H1299, H1975, H226) after TET1 knockdown. The knockdown of TET1 in lung cancer cell lines TET1 acquired a senescent phenotype defined by a change in morphology and p21/β-galactosidase upregulation as well as the percentage of micronucleated cells. Interestingly, depletion of WT-p53 expression or function sensitizes lung cancer cells to TET1 knockdown–induced cellular senescence [78]. PRC2 complex induced multiple features of cellular senescence in sensitive tumors. MAK683, a potent PRC2 inhibitor, in multiple sensitive cells (G401, G402, A2780, RD) induced a significant arrest cell cycle in G0/G1, increased p16 protein and SASP phenotype. In G401 xenograf, MAK683 treatment reduced Ki-67, increased SA-β-gal activity, and higher p16 levels. However, in xenografs observed increased expression of typical SASPs (GATA4, CCL2, IL-6, and IL-1B) [79]. Studies identified that BMI-1 protein, a component of the Polycomb Repressive Complex 1 (PRC1), is a critical factor for Diffuse Intrinsic Pontine Gliomas (DIPG), and its depletion significantly inhibited cancer cell proliferation and retarded tumor xenograft growth. In SF8628 cells, inhibition of BMI-1 through shBMI-1 or two inhibitors of BMI-1 (PTC-028, PTC-209) increased the level of SA–ß-gal. However, in DIPG cells, suppression of BMI-1 increased the expression of some senescence markers (p19, p21, GLP-1) and secreted SAPS factors (Il-6, Il-8, VEGF, CXCLS, CTGF). The data strongly suggested that BMI-1 could directly regulate SAPS factors by binding directly to promoters [80].
SEDT1A is a methyltransferase that, through promoter H3K4 methylation, regulates several genes that orchestrate mitosis and DNA damage responses. It was observed that SKP2 is a significant target of SETD1A and regulates the turnover of two cell cycle regulators (p21, p27). The elimination of SETD1A knockdown in breast cancer cells expressed characteristic SA-β-gal activity, increased cytokine and chemokine activity consistent with the SAPS phenotype, and the cell cycle arrest by reducing the expression of p27 and p21 [81].
Another H3K4 methyltransferase is JARID1B. Ohta et al. (2013) showed that JARID1B could be a molecular target for therapy-resistant cancer cells by inducing cellular senescence. In colorectal cancer Colo201 cells, shJARID1B induced cellular senescence through increased SA-β-gal activity and phosphorylation of Jnk/Sapk, as well as higher intracellular ROS levels [82].
Other enzymes involved in epigenetic modifications are sirtuins, essential factors that delay cellular senescence and extend the organismal lifespan. Three mammalian sirtuins are located within the mitochondria (SIRT3, SIRT4, and SIRT5), while the other sirtuins exert their functions within the cytosol (SIRT2), in the cell nucleus (SIRT1 and SIRT6) or within the nucleoli (SIRT7) [83]. Recently, studies have shown that sirtuins are involved in cancer cell senescence. SIRT1 is a nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase that plays an important role in cancer development, progression, and therapeutic resistance, making it a viable therapeutic target. Knockdown of SIRT1 (SIRT1-KD) in prostate cancer cells decreased the expression of negative regulators of senescence, including CENPA, EXO1, E2F1, increased expression of p21 (CDKN1A) under androgen deprivation conditions, and increased activity and levels of SA-β-galactosidase. However, it was suggested that SIRT1 silencing prevents prostate tumor growth, possibly by inducing senescence. The silencing of SIRT1 combined with 0.5 Gy radiation effectively eliminated the colony formation of prostate adenocarcinoma cells, highlighting the potential for inhibition of SIRT1 in radio-sensitization [84].
Up-regulation of another sirtuin- SIRT2 is a specific feature associated with stress-induced premature senescence but not quiescence or cell death. Doxorubicin treatment of the osteosarcoma U2OS cell line influenced induction senescence by positive for senescence-associated β-galactosidase activity, enlarged and flattened morphology, increased expression levels of growth markers (p53, p21) and marker of the secretory phenotype (PAI-1), loss of nuclear membrane protein lamin B1. Doxorubicin in U2OS cells also caused an arrest that was accompanied by the G2 specific marker- cyclin B1. However, an increase in the protein level of both SIRT2 and SIRT4 was observed in doxorubicin-induced senescence cells compared to non-senescent control cells. An increase in SIRT2 levels could also be observed in doxorubicin-induced senescent cells such as A375 (a human melanoma cell line) and A549 cells (NSCLC cell line). An increase in SIRT2 levels during senescence is also accompanied by changes in the acetylation status of its downstream targets through the deacetylation of α-tubulin at lysine 40, histone H4 at lysine 16 (H4K16) and p65 at lysine 310. Pretreatment with N-acetyl cysteine (NAC) could effectively rescue U2OS cells from doxorubicin- induced senescence [85].
Furthermore, NAC treatment also attenuated SIRT2 accumulation due to stress-induced premature senescence. The p53-p21 axis plays an important role in the initiation of premature senescence. Suppression of p53 largely prevented the expression of doxorubicin-induced SIRT2 in U2OS cells. Treatment of U2OS cells with nutlin stabilized not only p53 levels but also induced premature senescence, which was accompanied by increased levels of SIRT2. Furthermore, combined treatment with doxorubicin and nutlin not only accelerated senescence but also showed higher levels of SIRT2 compared to treatment with nutlin or doxorubicin alone. These results showed that the increase in SIRT2 expression in senescent cells depends on p53 status [85].
Mitochondrial sirtuin SIRT3 is significantly overexpressed in multiple human melanoma cells. In melanoma cell lines (SK-MEL2, G361, SK-MEL-29), SIRT3 knockdown induces senescence-like phenotype. SIRT3 knockdown altered cell morphology, showing a somewhat irregular, enlarged, flat, and multinucleated phenotype. After SIRT3 knockdown, changes in molecular markers of senescence were also observed, such as increase in SA-β-gal activity and SAHF formation, up-regulation of levels of p16INK4a and p21Waf1, enhanced accumulation of cells in phase G0 / G1, decreases in expression of cyclins (D1, E1) and Cdks (2, 4, 6) [86].
Mitochondrial sirtuin SIRT4 has been shown to play a tumor-suppressive role in many cancers, including hepatocellular carcinoma (HCC). In hepatitis B virus-related hepatocellular carcinoma, (HepG2-HBx, HepG2.2.15 cells), SIRT4 overexpression showed that they became large, flat, granular, and the majority of cells, an increased percentage of β-galactosidase positive cells, an increase in p16 and p21 expression, decreased expression of cyclin-dependent kinase (cyclin B1, cdc2, cdc25c) and survivin [87].
SIRT6, one of the sirtuins, regulates DNA repair, cell metabolism, and transcription by targeting various substrates for deacetylation or ADP-ribosylation. Studies showed that silencing of SIRT6 could induce hepatocellular carcinoma cell senescence. The SIRT6-silencedHep3B and Huh-7 cells were the characteristic morphology of cellular senescence cells. In SIRT6-depleted HCC cells positively stained with SA-ß-gal and mRNA level of IL8, CXCL1, CXCL2 and CXCL3 were increased. However, induction of cellular senescence in HCC cells by SIRT6 depletion in p16 / Rb and p53/p21- independent manners. SIRT6 silencing increased cyclin B1 and downregulated cyclin E expression as well as elevated levels of phosphorylation of CDC2 on Tyr15. Furthermore, the fraction of BrdU-positive cells that proliferated decreased among SIRT6-depleted cells, indicating that SIRT6-depleted HCC cells were arrested in the G2/M phase [88].
miRNAs are evolutionary conserved short noncoding RNA molecules that down-regulate the expression of their mRNA targets. These small molecules could regulate a host of cellular processes. Dysregulation in miRNA expression has been shown to modulate the pathological pathways involved in the development of various diseases, such as diabetes, cancer, and CVS. miRNA could be used as molecular markers in a normal physiological and disease state [89]. Recent reports have indicated that miRNAs act as key modulators of cancer cellular senescence by targeting critical regulators of senescence pathways [90].
miR-449a is located on chromosome 5 and shares seed sequences with miR-34a, a known tumor suppressor. Increasing evidence has shown that miR-449a is often underexpressed in several types of malignant tumors, including the prostate, gastric, and bladder. In these types of cancer, miR-449a induces cell cycle arrest, apoptosis, and senescence by regulation of critical cell cycle motors or apoptosis inhibitors, including histone deacetylase-1 (HDAC-1), CDK6, CDC25A, or sirtuin 1 (SIRT1). In lung cancer A549 and 95D cells, miR-449a mimics caused significant G0/G1 arrest, positive staining for senescence-associated β-galactosidase, and changes in cell morphology such as increased size and a broad, flattened shape. These results suggest that miR-449a suppresses cell proliferation and induces cell senescence [91].
Epstein–Barr virus–associated gastric cancer (EBVaGC) is approximately for about 10% of all gastric cancer cases and has unique pathological and molecular characteristics. EBV encodes many microRNAs that actively participate in the development of EBV-related tumors. It was reported that EBV-miR-BART3-3p (BART3-3p) promotes the growth of gastric cancer cells in vitro and in vivo. The BART3-3p encoded by EBV regulates the cellular senescence pathway in gastric cancer. BART3-3p plays a negative role in regulating p53/p21 in EBVaGC. It inhibits the expression of the PTEN protein, as well as some cytokines (IL-1A, IL-1B, IL-6 and IL-8), thus preventing the senescence of GC cells and promoting the progression of EBVaGC [92].
miR-21 has been reported as an oncogene in PCa, and its expression level was associated with chemotherapy-resistant CRPC. Additionally, elevation of miR-21 has been detected in several human cancers, including breast, gastric, colon, lung, pancreatic, and ovarian cancers with a poor prognosis. miR-21 deficiency suppresses prostate tumorigenesis by downregulating the IRS1/SREBP-1 signaling pathway and inducing cellular senescence. In prostate tumorigenesis of Pten/Trp53 mutant mice, miR-21 deficiency decreased levels of IRS1, SREBP-1, FASN, ACC, pAKT, Ki67, β-galactosidase and decreased nuclear SREBP-1 [93].
miR-137 levels are significantly reduced in human pancreatic tumors, consistent with previous studies revealing a defective senescence response in this type of cancer. Restoration of miR-137 expression in pancreatic tumor PANC-1 cells can trigger cellular senescence. In these cells, miR-137 significantly decreased growth rate and increased β-galactosidase activity. Comparable results were obtained with siRNA-mediated knockdown of KDM4A in PANC-1 cells, suggesting the possible contribution of KDM4A depletion to trigger senescence in PANC-1 cells. Studies showed that miR-137-induced senescence can be restored in PANC-1, thus blocking its proliferation [94].
High expression of miR-433 is significantly associated with poor progression-free survival (PFS) in patients with HGSOC and demonstrated that down-regulation of the mitotic arrest deficiency protein MAD2 by miR-433 induced cell chemoresistance to paclitaxel. Stable expression of miR-433 in ovarian cancer cells induced cellular senescence through decreased p-Rb, CDK6 and increased β-gal activity and the level of Il-6 and Il-8 cytokines [95].
The role of miR-494-3p in carcinogenesis appears to depend on the type of cancer. The tumor suppression effect of miR-494-3p has been reported in pancreatic cancer, cervical cancer, small cell lung cancer, and OSCC. The overexpression of miR-494-3p could induce cellular senescence in SAS cells by increasing p16INK4a, p21, p53, and β-gal. Studies showed that BMI-1 is a potential target for miR-494-3p. In SAS cells transduced with sh-Bmi1, lentivirus-transduced SAS cells activated the cellular senescence pathway. The radiosensitivity of SAS cells was increased at the radiation dose of 8 Gy. The overexpression of Bmi1 with a pcDNA3-Bmi1 vector in SAS cells showed a partial recovery of the radiosensitization effect of the miR-494-3p mimic. Studies suggested that miR-494-3p could improve radiosensivity in SAS cells by inducing cell senescence caused by downregulation of Bmi1 [96].
miR-22 is up-regulated in human senescent fibroblasts and epithelial cells but down-regulated in various cancer cell lines. Overexpression of miR-22 induces suppression of growth and acquisition of a senescent phenotype in normal and cancer cells. miR-22 caused notable and characteristic morphological alterations, including enlarged cellular size and flattened shape, and significantly increased SA-β-gal activity, cell arrest in the G1 phase, and inhibited the growth of in the breast cancer cell line. SIRT1 and CDK6 are genes involved in cell senescence, possibly through the pRb pathway. The overexpression of miR-22 markedly decreased SIRT1, SP1, and CDK6 in SiHa and MDA-D3 cells by binding to their 3′-UTR promoter regions [97].
miR-485-5p is located at a fragile site on chromosome 14q32 and abnormal expression of miR-485-5p was observed in many types of malignant tumors such as ovarian cancer, non-small cell lung cancer, hepatocellular carcinoma, gastric cancer, ependymoma, glioma, oral tongue squamous cell carcinoma and melanoma [98]. The binding of miR-485 5p to CDK16's 3'-UTR promotes senescence. In lung cancer cells, miR-485-5p overexpression resulted in senescence‐associated phenotypes, including a reduced cell proliferation rate, an increased percentage of positive SA‐β‐gal‐staining cells, and cells arrested in the G1 cell cycle phase [57].
miR-7, which acts as a tumor suppressor in various types of cancer, including pancreatic cancer, can down-regulate poly (ADP-ribose) polymerase 1 (PARP1), which functions as a regulator of various biological processes, including DNA repair and chromatin remodelling in the senescence programme. It is well known that gemcitabine is a first-line chemotherapeutic agent in pancreatic cancer. Gemcitabine induced senescence in PANC-1 cells, measured by decreased cell proliferation, increased SA-β-gal activity, elevated senescence-associated signaling, and increased expression levels of several SASP factors, mainly dose-dependent. miR-7 may be involved in resistance togemcitabine through the regulation of senescence. miR-7 regulate the PARP1/NF-κB signaling and negatively regulated H3K9me3 formation. miR-7 overexpression resulted in a marked decrease in cellular PARP1 and subsequent NF-κB inactivation, further repressing the senescence-associated phenotype. The overexpression of miR-7 in gemcitabine-resistant PANC-1 cells markedly decreased cellular senescence, including decreased SASP SA-β-gal activity, decreased expression levels, and down-regulation of SASP and downregulation of PARP1/NF-κB senescent signaling [99].
miR-34a, which can function as a tumor suppressor, is deregulated in many human cancers, including NSCLC. Recent studies have demonstrated that miR-34a modulates IR-induced senescence in NSCLC. miR-34a overexpression enhances IR-induced senescence in NSCLC cells by targeting c-Myc. miR-34a matches precisely the 3’-untanslated region (3’-UTR) sequence of the MYC oncogene [70]. miR-34a was highly up-regulated in a human colon cancer cell line, HCT 116. The introduction of miR-34a into HCT 116 cells caused senescence-like phenotypes with positive staining for senescence-associated β-galactosidase (SA-β-gal), through downregulation of E2F and up-regulation of p53 / p21, increased cell size and decreased cell proliferation [100].
miR-130b~301b cluster shows tumor suppressor functions in vitro, influencing cell cycle, cell viability, apoptosis, and invasion. Transfection of miR-130b or miR-301b was associated with significant up-regulation of the tumor suppressor genes CDKN2A (p16) and, more dramatically, CDKN2B (p15), along with downregulation of LMNB1, increased β-galactosidase (GLB1) mRNA levels, decrease in H3K9me3 and MMP1, MMP10 and CCL20 expression, along with overexpression of IL-1A, IL-1B and IL-6. The overexpression of miR-130b or miR-301b resulted in the arrest of cells in phase S, and the overexpression of miR-130b arrested cells in G2 / M [101].
The up-regulated miR-203 level was related to unsatisfactory clinical outcomes and a high probability of carcinogenesis and recurrence in human malignancies, including hepatocellular cancer, glioma, melanoma, gastric cancer, breast cancer, and CRC. Studies also demonstrate that miR-203 acts as a tumor suppressor in tumor progression [102]. Ectopic expression of miR-203, which functions as an anti-oncomir and its downregulation, inhibits cell growth in canine oral malignant melanoma tissue specimens, as well as in canine and human malignant melanoma cells. Studies showed that miR-203 could also induce senescence in human melanoma cells. Cells transfected with miR-203 have shown a decrease in the number of cells positive for SA-β-gal activity, SIRT1. Furthermore, the ectopic expression of miR-203 in Mewo cells significantly and dose-dependently decreased the expression levels of the E2F3a, E2F3b, and ZBP-89 proteins, resulting in cell cycle arrest [103] [Fig. 1, Table 4].
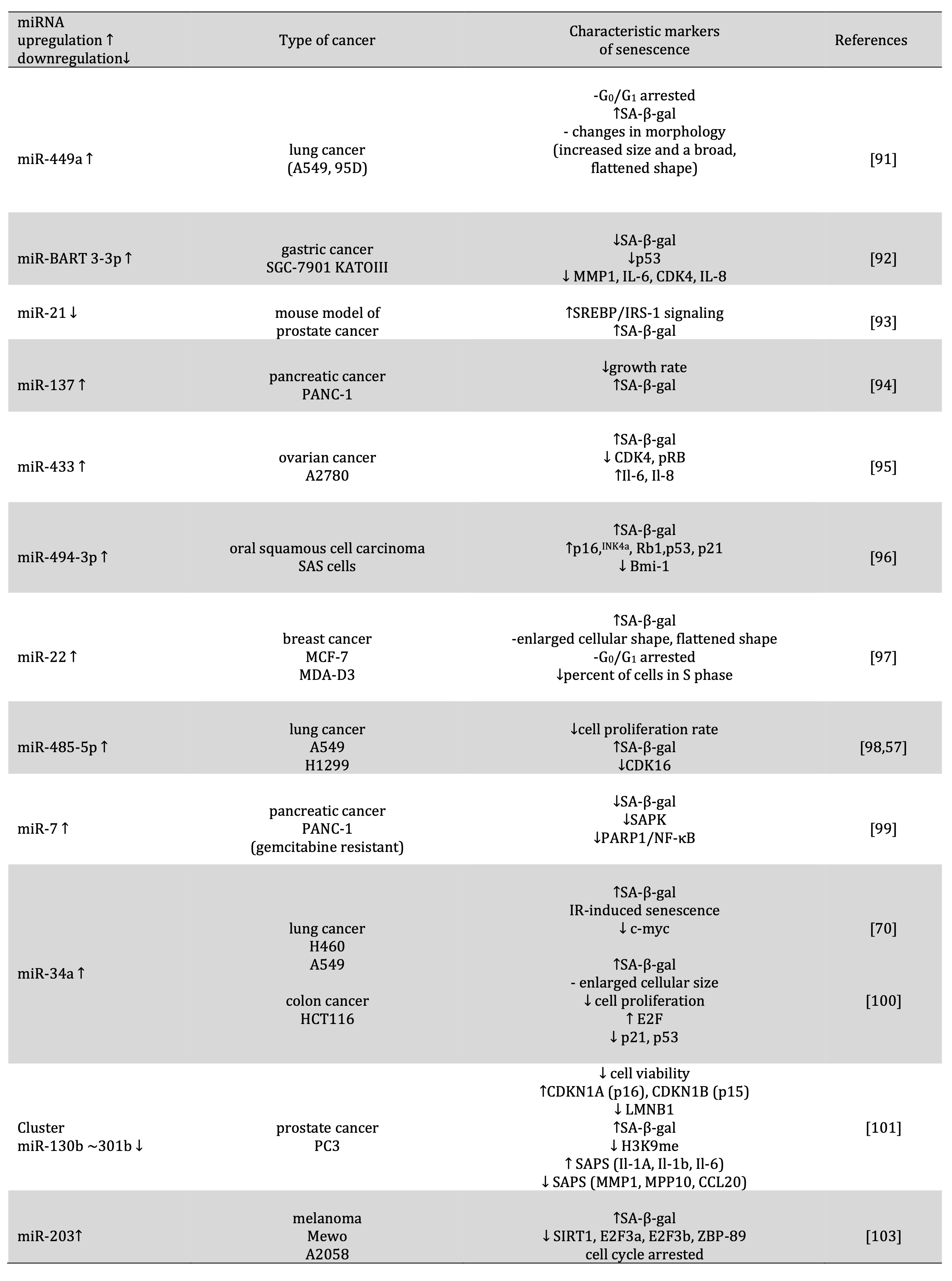
Table 4: Role of miRNA in the induction of cancer cell senescence
Two faces of cancer cell senescence. Positive and negative aspects of senescence on cancer progression
Senescence is generally regarded as a tumor-suppressive process by preventing cancer cell proliferation and suppressing malignant progression from pre-malignant to malignant disease. It is suggested that senescence is one of the obstacles to oncogenesis because cell cycle arrest and inhibition of cell proliferation were observed [104]. Cell progression regulation depends on cyclin-dependent kinases (CDKs) and their regulatory cyclin subunits. The molecular mechanisms of cells arrested in phase involve CDK inhibitors such as p21, p15, p16, and p27, whose expression is upregulated in senescent cells. Cancer cells divide more rapidly than normal cells. This is manifested by altered expression and/or activity of cell cycle-related proteins, growth factors, and changes in signal transduction pathways that also stimulate cell growth. EGFR is one of the receptor tyrosine kinases important in controlling cell proliferation. In some types of cancers, we observed an increased level of this protein and, as a result, higher cell proliferation. In gallbladder cancer cells, down-regulation of EGFR induced cancer senescence and inhibited cell proliferation by up-regulation of p21 and induction cell cycle arrested [55]. However, in lung cancer cells, the knockdown of the cyclin-dependent kinase CDK16 could induce senescence and decrease the proliferation rate of cancer cells. Thus, induction of senescence through down-regulation and expression of some proteins involved in theregulation of the cell cycle in cancer cells may play an important role in cancer cell proliferation and progression [57]. Recently, data showed that some types of cells in the tumor microenvironment, through the secretion of various factors, influenced cancer cell senescence. Cells in the tumor microenvironment produce a bioactive secretome, termed the senescence-associated secretory phenotype (SASP). One of the components of the tumor microenvironment is cancer-associated fibroblasts, macrophages, or other autoimmune cells that secrete a variety of soluble signaling factors, such as chemokines and cytokines. Some SASP factors reportedly play an essential role in the onset of stable cell‐cycle arrest in senescent cells, presumably contributing to the tumor suppressing function of cellular senescence. SAPS-induced or enhanced senescent-associated growth arrest in autocrine and paracrine manners through production through a pro-inflammatory environment, recruitment of immune cells, and inhibition of cancer progression. The role of paracrine and autocrine senescence in cancer is poorly investigated [3]. Inflammasome-mediated IL-1 is a crucial regulator of senescence and controls the activation of the SAPS programme. In mouse models of pancreatic cancer, genetic inactivation of IL-1α could recapitulate delayed cancer progression. These studies suggested that Il-1, as an inflammatory component of SAPS in an autocrine manner, influences tumor suppression [105]. Some components of SAPS (Il-8, Il-6, PAI-1, IGHBP7) reinforce senescence growth arrest in vitro [106, 107]. It was observed that SASP could contribute to an antitumor microenvironment by skewing macrophage polarization to a tumor-inhibiting M1 state in a fibrosis-associated liver cancer model [108].
Moreover, cells co-cultured with senescent cells showed a phenotype similar to senescent cells. Furthermore, senescent cells induce paracrine senescence in neighbouring cells through SASP, which acts as a barrier against tumor growth [109]. SASP can also activate immune surveillance by p53 and up-regulation of some cytokines. The immune system plays an important role in tumor surveillance. In malignant hepatocytes, induction of senescence by p53 contributed to tumor clearance through SAPS-mediated differentiation and up-regulation of inflammatory cytokines. These processes lead to cell cycle arrest in vitro, but in vivo they trigger an innate immune response against tumor cells [110].
Paradoxically, senescent cells are frequently observed in cancer tissue but have a critical anti-tumor barrier. Through autocrine secretion of chemicals and cytokines, pre-malignant senescent hepatocytes gave a signal from liver-infiltrating immune cells to tumor clearance. In this senescence surveillance, antigen-specific CD4 (+) T cells are involved, natural killers that could eliminate senescent tumor cells and consequently suppress tumorigenesis [108, 111, 112].
Studies confirm that senescence can be a barrier to tumor development, but there is growing evidence that senescent cell accumulation cooperates in cancer progression. Some studies showed that senescent fibroblasts in the microenvironment could promote epithelial cell growth and tumorigenesis. Krtolica et al. (2001) observed that senescent fibroblast co-cultured with preneoplastic epithelial cells (HaCAT, S1, SCp2) and neoplastic epithelial cells (MDA-MB-231, HT1080, SOAS-2) stimulated the growth of these cells. The ability of senescent fibroblast to promote cancer progression confirmed a study showing that co-cultured senescent fibroblast with premalignant breast epithelial cells irreversibly lose differentiation characteristics, gain invasiveness, and undergo malignant transformation of premalignant breast epithelial cells [113]. Senescent fibroblasts also strongly accelerate tumorigenesis in in vivo mice models [33]. It was observed that senescent fibroblasts could stimulate epithelial cell proliferation, migration, and invasion by secreting MMP-3 [113], Il-8 overexpression [114], VEGF secretion, and increase little HIF-1 α [115].
Another component that promotes tumor progression is SAPS. Some studies confirmed that SAPS suppresses tumor formation, but there is evidence that these components could promote cancer development. SAPS created an immunosuppressive environment and contributed to tumor progression and relapse. Il-6 and Il-8 are components of SAPS, which could play a role in stimulating inflammation, epithelial-to-mesenchymal transition (EMT), and invasion [113]. In a mouse model that mimics the aged skin microenvironment, senescent stromal cells contributed to tumor promotion through the secretion of IL-6, increasing the number of myeloid-derived suppressor cells (MDSCs) and increasing the ability of MDSCs to inhibit anti-tumor T-cell responses [116]. Another study suggested that senescent mesenchymal stem cell xenograft mice, activating the IL-6 / STAT-3 signalling pathway, stimulate the proliferation and migration of breast cancer cells in vitro and promote tumor progression [117-119]. Studies showed that IL-6 could cooperate with HER-2 and promote tumor development. It has been shown that in HER-2-positive breast cancer, approximately 5% of tumor cells constitute senescent cells. Blocking IL-6 inhibited tumor growth, indicating that IL-6 was secreted through senescent cells, promoting cancer progression [120-121]. In the in vitro dormancy model of MCF-7 breast cancer cells, bone marrow stroma secretory senescence released the IL-6, IL-8, and TGFβ1 components reactivated dormant MCF-7 cells, promoting their mesenchymal appearance and, as a result, increasing cellular proliferation and migration [122]. The next cytokine secreted by both cancer and noncancer cells, which creates a pro-inflammatory, microenvironment is Il-1. This cytokine has been shown to induce cells into “pseudo-senescence.” Under chronic IL-1, cancer cells could be drug-resistant in this state, acquire stem-like properties and consequently drive cancer recurrence [123].
Another factor secreted by senescent cells is MCP-1 chemokine. MCP-1 recruited immune cells expressing the CCR2-receptor [106, 107]. In hepatocellular carcinoma, CCR2-positive myeloid cells recruited by senescence improved growth and were correlated with patients with a worse prognosis with hepatocellular carcinoma through inhibition of NK cells [124].
Studies showed that cisplatin could induce senescence in melanoma cells and xenograft mice [122]. Cisplatin in melanoma cells induced the release of IL-8 and IL-1α, which activated the ERK1 / 2-RSK1 pathway and promote the growth of non-senescent cells. Acceleration of tumor growth was observed after transplantation of non-senescent and senescent melanoma cells compared totransplantation of non-senescent cells only in mice. However, only senescent cells do not produce tumors [122].
Recently, studies showed that treatment with the same dose of chemotherapy lymphomas resulted in a higher tumor-initiating potential of previous senescent cells than never senescent cells. Furthermore, senescent cells previously retained the ability to re-enter therapy-induced senescence (TIS) after re-exposing to chemotherapy [125]. These results have been shown to depend on Wnt signaling, which was activated in TIS. Wnt signaling is essential for stem cell renewal and enhanced tumor initiation capacity [125-127]. Tumor induces senescence by inducing cancer stem cells contributed by chemo-resistance [128]. Cell senescence could affect the promotion of cell-autonomous reprogramming of non-stem cancer cells into cancer stem cells [129, 125]. The important mediators of senescence-induced cell reprogramming are the transcription factors Oct4, Sox2, Klf4, c-Myc (OSKM), and IL-6, while expression could depend on the absence of loci INK4a and p53 [65, 129-131].
The important role in senescent cells is playing the role of signaling molecules called tumor suppressors (p53, c-myc, PTEN). These molecules could induce tumor invasiveness [132, 125, 133]. Recent studies have shown that the c-myc factor could affect cell senescence and the cancer mechanism of sustained tumor regression of haematopoietic tumors, osteosarcomas, and hepatocellular carcinomas. Afterthe inactivation of c-myc, several molecular characteristics of cellular senescence were observed, such as elevated β-gal activity, increased expression of cell cycle inhibitors p15INK4b and p16INK4a, the formation of heterochromatin, and methylation of H3K9. However, in studies, some tumor suppressors (p53, Rb, and p16INK4a) play an essential role in regulating c-myc and inducing cell senescence through these transcription factors. Lymphoma cell lines expressing endogenous or restored p53 exhibited SA-β-gal activity after inactivation ofMYC [43].
Furthermore, p53 expression was required for complete tumor regression after MYC inactivation when transplanted into syngeneic hosts. In osteosarcoma cells, after suppression of Rb, p16INK4a, or p53, the loss of SA-β-gal staining was observed after MYC inactivation and continued cellular proliferation. In osteosarcoma, inactivation of c-myc and shRNA against p16INK4a or Rb exhibited sustained regression [43]. Recently, studies have shown that senescence plays a key tumor suppressor role in Pten-deficient prostates. In the mouse model, complete loss of PTEN, a strong p53-dependent senescence response, could oppose tumor progression. Total loss of PTEN induction of senescence and inhibiting tumor progression as well as leading to metastasis. Recent studies showed that inactivated mutation of some genes (SMAD4, CyclinD1, SPP1, p53, Nkx3.1), as well as those overexpressing Erg, HER receptors in the null mouse model, overcame senescence, leading to aggressive metastatic CaP [134]. However, therapies based on PICS induction can lead to replication stress and, consequently, to additional mutations, allowing escape from senescence, cancer progression, and induction of the secretion of cytokines and chemokines contributed to tumor development. Interestingly, in PTEN-deficient prostatic epithelial cells, ablation of p53 allowed avoidance of the senescence barrier to promote development and invasiveness [135].
Treatment modalities used in cancer and their connection with cell senescence
Therefore, senescence could promote tissue dysfunction and the early onset of various age-related symptoms in treated cancer patients. Studies indicate that the presence of numerous therapies or compounds used in cancer treatment could related to senescence. It turns out that cancer cell senescence could be related to chemotherapy, radiotherapy, CDK4/6 inhibitors, epigenetic modulators, immunotherapy, natural compounds, and hormonal therapy using [7, 136] [Fig. 2]
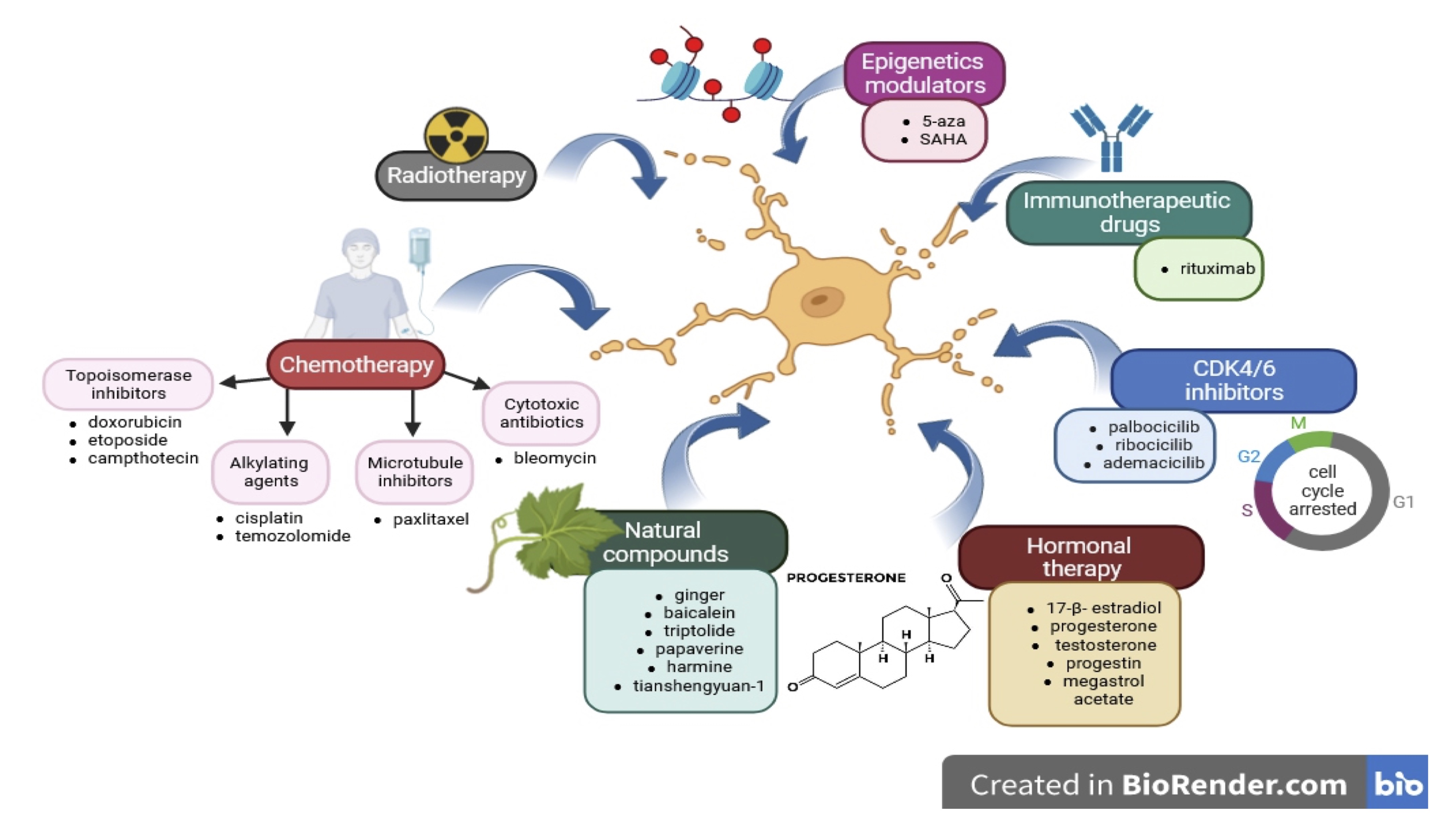
Fig. 2: Treatment modalities used in cancer and their connection with cell senescence. Cancer cell senescence could be related in chemotherapy (topisomerase inhibitors, alkylating agents, microtubule inhibitors, cytotoxics antibiotics), radiotherapy, CDK4/6 inhibitors (palbocicilib, ribococicilib, ademacicilib), epigenetic modulators (5-aza, SAHA), immunotherapy (rituximab), natural compounds (ginger, balcalein, triptolide, papaverine, harmine, tianshengyan-1) and hormonal therapy (17-β-estradiol, progesterone, testosterone, progestin, megestrol acetate.
Chemotherapy
In chemotherapy, commonly used topoisomerase inhibitors (doxorubicin, etoposide, camptothecin), alkylating agents (cisplatin, temozolomide), microtubule inhibitors (paclitaxel), cytotoxic antibiotics (bleomycin) could be related in cancer cell senescence [109].
Topoisomerase inhibitors are compounds that block topoisomerase enzymes and prevent the replication process. The most common topoisomerase inhibitor used in chemotherapy of various cancers, including lung, breast, lymphoma, and lymphocytic leukaemia, is doxorubicin [137]. Studies showed that doxorubicin induces senescence in some types of cancers, including fibrosarcoma (HT1080), breast cancer (MCF-7, MDA-MB231), colon cancer (HCT116), prostate cancer (PC3, LNCaP), ovarian cancer (A2780), glioma (U251), leukaemia (K562), melanoma (SK-MEL-103) in low doses of doxorubicin in the range of 20-100 nM and within 2-4 days. Another topoisomerase inhibitor-induced cancer cell senescence is etoposide. The etoposide-induced senescence fibrosarcoma cell line HTC1080 is characterised by elevated β-gal activity, arrest cells of the G1 phase, and increased micronuclei formation. Simultaneous effects were observed after camptothecin treatment for colon cancer (HCT116) and lung cancer (H1299) [138, 139].
Alkylating drugs can lead to the formation of cross-links in the DNA molecule. The formation of cross-links inhibits or prevents transcription, DNA synthesis, and blocks cell division. The typical alkylating agents that can induce cancer cell senescence are cisplatin and temozolomide. Cisplatin could induce senescence in nasopharyngeal carcinoma (CNE1) and fibrosarcoma (CNE1) [139, 140], whiletemozolomide caused senescence in glioma [141-144], melanoma [145], colorectal cancer [146].
Paclitaxel is a microtubule inhibitor, which compromises the metaphase-anaphase transition, thus arresting the cell at mitosis. These inhibitors are commonly used in treating ovarian, breast, brain, and non-small cell lung cancer. In MCF-7 breast cancer cells, paclitaxel-induced senescence through reduction of cell proliferation, increasing DDR, and elevated SA-β-galactosidase activities [147-148].
Bleomycin is a drug commonly used in Hodgkin lymphoma and testicular germ therapy. This compound oxidizes DNA nucleotides induced single- and double-strand breaks of DNA [149]. Studies showed that in A549 lung cancer cells bleomycin induced senescence by upregulation of p16, p21 expression, and SA-β-galactosidase activities [150-152] [Fig 2].
Radiotherapy
Radiotherapy is one of the methods used in treating many cancers, including carcinomas (skin, head and neck, lung, breast, bladder and prostate, lymphoma, soft tissue sarcoma, and nervous system [69]. Studies suggested that radiotherapy could induce cancer cell senescence, and induction of senescence depends on p53 status and mutations affecting survival pathways. However, induction of senescence in cancer cells depends on the dose and time of exhibition of irradiation. Irradiation in 2 Gy to 10 Gy could induce senescence in breast cancer (MCF-7, MDA-MB-231) and glioblastoma cell lines (HT1080) [139, 153-155] [Fig. 2].
CDK4/6 inhibitors
One of the anticancer strategy is the critical inhibition factors for the cell cycle transition from G1 to S phase, CDK4, and CDK6 [156].Three CDK4/6 inhibitors are used in cancer therapy - palbociclib, ribociclib, and abemaciclib. Recently, studies have shown that CDK4/6 can also induce senescence in cancer cells [157-159]. Palbociclib induces the cancer senescence in the case of breast cancer (MCF-7, T48) [148-160], melanoma (Mel10, 1205Lu, SK-MEL-103) [161-164], liposarcoma (LS8817, LS141, LS0082, U2OS, LS8107) [165-167], gastric cancer (AGS, MKN-45) [168], hepatocellular carcinoma (Huh7, skHep1) [169]. In turn, ribocicline initiates senescence in Edwig's sarcoma cells (SKNEP1) and neuroblastoma cells [157-170], and abemaciclib in various types of breast cancer cells (MDA-MB-453, BT474, MCF-7). CDK4/6 inhibitors in cancer cells induced senescence by elevated SA-β-galactosidase activities, reduced cell proliferation, up-regulation of cyclins D1 and D3, p16, p21, and down-regulation of p-Rb, G1 growth arrest, SAPS induction, reduced BrdU incorporation [171-175] [Fig 2].
Epigenetic Modulators
Studies showed that some epigenetic modulators 5-Aza-2′deoxycytidine (5-aza) and suberoylanilide hydroxamic acid/ vorinostat (SAHA) induced cancer cell senescence. In osteosarcoma cells (U2OS), 5-aza upregulation of p16 and arrest of growth. Elevated SA-β-galactosidase activity and increased DDR after 5-aza treatment were observed in lung mesothelioma cells (H28). 5-aza induced senescence in U2OS and MCF-7 by activating the p53-p21 pathway [176-180]. Another epigenetic modulator that induces cancer cell senescence is SAHA. These compounds are histone deacetylase (HDAC) inhibitors and have been approved for treating cutaneous T-cell lymphoma. After SAHA treatment, some markers of senescence in leukaemia cells (MOLM-7, HL-60, and JURL-MK1), colon cancer cells (HCT116), and urothelial carcinoma cells [181-182] [Fig 2].
Immunotherapeutic drugs
In cancer therapy, some immunotherapeutic drugs could cause apoptosis of immune-mediated cancer cells. One of these compounds is rituximab, a CD20-targeting antibody. Rituximab is used to treat leukaemia and lymphoma. Recently, studies showed that treatment of human lymphoma cells (EHEB, RC-K8, SD1) could induce senescence through elevated SA-β-galactosidase activities, DDR and SAPS induction, as well as upregulation of p53 and p21 [183] [Fig 2].
Natural Compounds
Studies indicated that natural compounds belonging to phenolics (ginger, pterostilbene), flavonoids (baicalein), terpenoids (triptolide), alkaloids (papaverine, harmine), TCM extracts (tianshengyuan-1) induce senescence in cancer cells [136].
Ginger exerts antimicrobial, antitumor, anti-inflammatory activity, and resistance to diabetes. In A549 lung cancer cells, ginger suppressing hTERT expression and telomerase activity caused telomere shortening and triggering cell senescence [184-186].
Pterostilbene, similar to ginger, in lung cancer cells could induce cancer senescence by suppressing telomerase activity, induction of DDR, activation of ATM/ATR/p53/p21, and blocking S phase [187-189].
Baicalein, a phenolic flavonoid compound, exerts powerful anticancer effects in various cancers. It has been shown that baicalein could significantly inhibit tumorigenesis in vitro and in vivo. However, recent studies indicated that baicalein, down-regulation of hTERT, and regulation of the MAPK / ERK / p38 pathway could regulate colon cancer cell senescence [190-191].
Triptolide (TPL) is a diterpenoid that indicates anti-fertility, anti-tumor and anti-cytogenesis effects. TLP could increase the expression of p53 and p21Kip1 and block the cell cycle. In liver cancer, TLP, by inhibiting cyclin, activating AKT signaling, suppressing hTERT expression and telomerase activity, contributed to the acceleration of liver cancer senescence and inhibition of tumor growth [192-193].
Papaverine (PPV), as an alkaloid, has a wide bioactivity for clinical use. The PPV is used as a muscle relaxant. Studies showed that PPV inhibited telomerase by reducing the transcription level of hTERT induction of senescence in hepatocellular carcinoma HepG2 cells. However, low PPV treatment doses accelerate senescence and elevated levels of SA-β-galactosidase [194-195].
Harmine is a β-carboline alkaloid with an antimutagenic, antidepressant, and antiplatelet effect. In breast cancer cells MCF-7, harmine induced cellular senescence and arrested cell proliferation. In these cells, some senescence markers, such as elevated levels of SA-β-galactosidase, decreased telomerase activity, and overexpression of the p53 / p21 pathway [196-197].
Tianshengyuan-1 (TSY-1) is an agent used in bone marrow deficiency and stimulates telomerase activity. Interestingly, TSY-1 had an inhibitory effect on telomerase activity in leukaemia cells with inherently high telomerase activities and an activation effect on normal blood mononuclear and stem cells with low telomerase activities. Studies showed that TSY-1 in acute myeloid leukaemia suppressed telomerase activity, but in peripheral blood mononuclear cells (PBMC) and Hematopoietic stem cells (HSCs), TSY-1 increased telomerase activity. The regulation of TERT genes through TSY-1 is probably dependent on the methylation of this gene. Tianshengyuan-1 could inhibit telomerase activity, promote cell senescence, and consequently block tumor growth [198-200] [Fig 2].
Hormonal therapy
Evidence supports that sex hormones are essential in cancer progression toward invasiveness and metastatic spread [201]. The latest research indicates that natural or synthetic sex hormones may play an essential role in the development of senescence in cancer cells.
17β-estradiol (E2) is a sex hormone that induces tumorigenesis and increases the risk of breast and ovarian cancers. However, recent data indicated the role of E2 in breast cancer senescence. Studies by Bajbouj et al. (2020) showed that E2 treatment of MCF-7 and MDA-MB-231 breast cancer cells reduced expression of p53, increased expression of p21 and LC3, presence of senescence-associated heterochromatin foci and higher levels of SA-β-galactosidase activity [202].
Progesterone is a hormone that has an anti-tumor effect in some types of cancer. Treatment with a high dose of progesterone could reduce the growth of glioblastomas and prolong survival in malignant glioma xenografts. Atif et al. (2019) measured the accumulation of SA-β-gal in U87MG-luc cells after 3 days of repeated exposure to progesterone at doses 0, 20, 40, and 80 μM. They observed the highest accumulation of β-gal in mice treated with 80 μM of progesterone compared to control mice. These results suggested that high doses of progesterone could induce premature senescence in glioblastoma cells [203]. Progesterone in the Hec50 endometrial cancer cell line caused time-dependent inhibition of the cell cycle by up-regulation of cyclin-dependent kinase inhibitors (p21, p27). Progesterone in Hec50 cells induced a secretory phenotype in the presence of progesterone A (PRA) and, to a greater degree, in the presence of progesterone B (PRB) receptor. However, progesterone-induced replicative senescence through β-gal activity, particularly PRA presence, but to a much lesser extent by PRB [204].
Bajalovic et al. (2022) observed the opposite effect after treating breast cancer MCF-7 cells in the synthetic form of progesterone- progestin (R5020). These synthetic hormones induced replicative senescence in MCF-7, expressing high levels of PRB. R5020 in MCF-7 caused a reduction in CDK2 and CDK4 and decreased in S phase cells. Progestin in MCF-7PRB cells activated the NF-κB pathway by phosphorylating IκBα and p65 pathways, leading to increased expression of SASP, such as IL-1a, IL-1b, and IL-8 [205]. Another synthetic form of progesterone is megestrol acetate. Use of this compound in the treatment of endometrial cancer Ishikawa and HHUA cells induced senescence by reducing cell growth, accumulation of β-gal, and increased expression of p16, p21. The effect of megestrol acetate on endometrial cancer senescence probably depends on the FOXO1 pathway and PRB expression [206].
Testosterone is the primary male sex hormone, but testosterone levels are a risk factor or predictor of a cancer prognosis that remains unclear. Some studies have shown that low serum testosterone can increase the risk of prostate cancer. In postmenopausal women, a high level of circulating free testosterone is associated with a reduced risk of lung cancer. However, elevated serum testosterone levels increased the risk of breast cancer in premenopausal women [207]. Recently, data showed that testosterone could affect cancer cell senescence. In prostate cancer cell lines LnCaP and Cw22rv1, induction of pulsed testosterone treatment G0 / G1 arrested, elevated level of β-gal activity, increased expression of p27, p16, and decreased expression of Spk2, cyclin D1, CDK4, c-myc, pRb [208] [Fig 2].
Conclusion
Senescence in cancer cells can be induced in various ways and depends on many molecular aspects. Studies showed that some mechanisms related to the regulation of telomerase activity, some senescence-related proteins, DNA damage response, tumor microenvironment, and epigenetic changes can promote cancer cell senescence [3-6]. It turns out that many molecular factors can be significantly related to cancer cell senescence, which suggests the complexity of the entire process.
The role of cancer cell senescence in cancer progression is ambiguous because induction of senescence could inhibit or induce tumor progression. This phenomenon can be molecular-dependent and mainly depends on the mutation or status of some proteins in cancer cells and the tumor microenvironment [120, 121, 135]. Currently, research is underway to determine potential mechanisms and the role of senescence in cancer cells and its impact on cancer progression, metastasis, and invasiveness in different types of cancers. Based on recent research, it can be suggested that senescence does not necessarily inhibit the progression of cancer. Analysis of the molecular basis of the senescence in various types of cancer may contribute to understanding the diverse role of senescence in cancer progression.
Studies showed that some compounds and therapies used in cancer treatment can be related to cancer cell senescence. Elucidating mechanisms underlying senescence have important implications for understanding its role in cancer cells. However, this process needs to be better understood, but very interesting in terms of the possibility of its application.
Acknowledgements
Author Contributions
Conceptualization, A.Z., writing—original draft preparation, A.Z., S.K.; writing—
review and editing, A.Z., A.R. All authors have read and agreed to the published version of the manuscript.
The article was carried out as part of the Interdisciplinary Research Grant of the University of Lodz (IDUB B2311001000201.07).
Funding Sources
This research received no external funding.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Schmitt CA, Wang B, Demaria M: Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol 2022 ;19:619-636.
https://doi.org/10.1038/s41571-022-00668-4 |
| 2 | Molina-Serrano D, Kyriakou D, Kirmizis A: Histone Modifications as an Intersection Between Diet and Longevity. Front Genet 2019;10:192.
https://doi.org/10.3389/fgene.2019.00192 |
| 3 | Yang J, Liu M, Hong D, Zeng M, Zhang X: The Paradoxical Role of Cellular Senescence in Cancer. Front Cell Dev Biol 2021;9:722205.
https://doi.org/10.3389/fcell.2021.722205 |
| 4 | Yasuda T, Baba H, Ishimoto T: Cellular senescence in the tumor microenvironment and context-specific cancer treatment strategies. FEBS J 2023;290:1290-1302.
https://doi.org/10.1111/febs.16231 |
| 5 | Nagineni CN, Naz S, Choudhuri R, Chandramouli GVR, Krishna MC, Brender JR, Cook JA, Mitchell JB: Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells. Int J Mol Sci 2021;22:4835.
https://doi.org/10.3390/ijms22094835 |
| 6 | Bloniarz D, Adamczyk-Grochala J, Lewinska A, Wnuk M: The lack of functional DNMT2/TRDMT1 gene modulates cancer cell responses during drug-induced senescence. Aging (Albany NY) 2021;13:15833-15874.
https://doi.org/10.18632/aging.203203 |
| 7 | Wang B, Kohli J, Demaria M: Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020 ;6:838-857.
https://doi.org/10.1016/j.trecan.2020.05.004 |
| 8 | Shay JW and Wright WE: Telomerase activity in human cancer. Cur Opin Oncol 1996 ; 8:66-71.
https://doi.org/10.1097/00001622-199601000-00012 |
| 9 | Razgonova MP, Zakharenko AM, Golokhvast KS, Thanasoula M, Sarandi E, Nikolouzakis K, Fragkiadaki P, Tsoukalas D, Spandidos DA, Tsatsakis A: Telomerase and telomeres in aging theory and chronographic aging theory (Review). Mol Med Rep 2020 ;22:1679-1694.
https://doi.org/10.3892/mmr.2020.11274 |
| 10 | Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA: Inhibition of telomerase limits the growth of human cancer cells. Nat Med 1999;5:1164-1170.
https://doi.org/10.1038/13495 |
| 11 | Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T: Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 2005;7:25-37.
https://doi.org/10.1016/j.ccr.2004.11.021 |
| 12 | Seimiya H, Oh-hara T, Suzuki T, Naasani I, Shimazaki T, Tsuchiya K, Tsuruo T: Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991. Mol Cancer Ther 2002;1:657-665.
|
| 13 | Okamoto K, Seimiya H: Revisiting Telomere Shortening in Cancer. Cells 2019 ;8:107.
https://doi.org/10.3390/cells8020107 |
| 14 | Shammas MA, Koley H, Batchu RB, Bertheau RC, Protopopov A, Munshi NC, Goyal RK: Telomerase inhibition by siRNA causes senescence and apoptosis in Barrett's adenocarcinoma cells: mechanism and therapeutic potential. Mol Cancer 2005;4:24.
https://doi.org/10.1186/1476-4598-4-24 |
| 15 | Lex K, Maia Gil M, Lopes-Bastos B, Figueira M, Marzullo M, Giannetti K, Carvalho T, Ferreira MG: Telomere shortening produces an inflammatory environment that increases tumor incidence in zebrafish. Proc Natl Acad Sci U S A 2020;117:15066-15074.
https://doi.org/10.1073/pnas.1920049117 |
| 16 | Fumagalli M, Rossiello F, Mondello C, d'Adda di Fagagna F: Stable cellular senescence is associated with persistent DDR activation. PLoS One 2014;9:e110969.
https://doi.org/10.1371/journal.pone.0110969 |
| 17 | d'Adda di Fagagna, F, Reaper, PM, Clay-Farrace, L, Fiegler, H, Carr, P, Von Zglinicki, T, Saretzki G, Carter NP, Jackson SP:DNA damage checkpoint response in telomere-initiated senescence.Nature 2003;426:194-198.
https://doi.org/10.1038/nature02118 |
| 18 | Lou Z, Chini CC, Minter-Dykhouse K, Chen J: Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J Biol Chem 2003;278:13599-13602.
https://doi.org/10.1074/jbc.C300060200 |
| 19 | Shiloh Y: ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3:155-168.
https://doi.org/10.1038/nrc1011 |
| 20 | Stein GH, Drullinger LF, Soulard A, and Dulic, V: Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 1999;19:2109-2117.
https://doi.org/10.1128/MCB.19.3.2109 |
| 21 | Beausejour CM, Krtolica, A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J: Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003;22:4212-4222.
https://doi.org/10.1093/emboj/cdg417 |
| 22 | Alster O, Bielak-Zmijewska A, Mosieniak G, Moreno-Villanueva M, Dudka-Ruszkowska W, Wojtala A, Kusio-Kobiałka M, Korwek Z, Burkle A, Piwocka K, Siwicki JK, Sikora E: The role of nibrin in doxorubicin-induced apoptosis and cell senescence in Nijmegen Breakage Syndrome patients lymphocytes. PLoS One 2014;9:e104964.
https://doi.org/10.1371/journal.pone.0104964 |
| 23 | Rouibah H, Kebsa W, Lahouel M, Zihlif M, Ahram M, Aburmaileh B, Mansour Al Shhab MA, Al-Ameer HJ, Mustafa E: Algerian propolis: between protection of normal cells and potentialisation of the anticancer effects of doxorubicin against breast cancer cells via P-glycoprotein inhibition and cell cycle arrest in the S phase. J Physiol Pharmacol 2021;72:2.
|
| 24 | Hu X, Zhang H: Doxorubicin-Induced Cancer Cell Senescence Shows a Time Delay Effect and Is Inhibited by Epithelial-Mesenchymal Transition (EMT). Med Sci Monit 2019;25:3617-3623.
https://doi.org/10.12659/MSM.914295 |
| 25 | Srdic-Rajic T, Santibañez JF, Kanjer K, Tisma-Miletic N, Cavic M, Galun D, Jevric M, Kardum N, Konic-Ristic A, Zoranovic T: Iscador Qu inhibits doxorubicin-induced senescence of MCF7 cells. Sci Rep 2017;7:3763.
https://doi.org/10.1038/s41598-017-03898-0 |
| 26 | Karabicici M, Alptekin S, Karagonlat ZF, Erdal E: Doxorubicin‐induced senescence promotes stemness and tumorigenicity in EpCAM−/CD133− nonstem cell population in hepatocellular carcinoma cell line, HuH‐7. Mol Oncol 2021;15:2185-2202.
https://doi.org/10.1002/1878-0261.12916 |
| 27 | Luo H, Yang A, Schulte BA, Wargovich MJ, Wang GY: Resveratrol induces premature senescence in lung cancer cells via ROS-mediated DNA damage. PLoS One 2013;8:e60065.
https://doi.org/10.1371/journal.pone.0060065 |
| 28 | Wang S, Wang Q, Wang H, Qin C, Cui X, Li L, Liu Y, Chang H: Induction of ROS and DNA damage-dependent senescence by icaritin contributes to its antitumor activity in hepatocellular carcinoma cells. Pharm Biol 2019;57:424-431.
https://doi.org/10.1080/13880209.2019.1628073 |
| 29 | Liu H, Zhao H, Sun Y: Tumor microenvironment and cellular senescence: Understanding therapeutic resistance and harnessing strategies. Semin Cancer Biol 2021;86:769-781.
https://doi.org/10.1016/j.semcancer.2021.11.004 |
| 30 | Xing F, Saidou J, Watabe K: Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed) 2010;15:166-179.
https://doi.org/10.2741/3613 |
| 31 | Ren C, Cheng X, Lu B, Yang G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment: Eur J Cancer 2013;49:3889-3899.
https://doi.org/10.1016/j.ejca.2013.07.140 |
| 32 | Dean JP, Nelson PS: Profiling influence of senescent and aged fibroblasts on prostate carcinogenesis. Br J Cancer 2008;98: 245-249.
https://doi.org/10.1038/sj.bjc.6604087 |
| 33 | Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J: Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A 2001;98:12072-12077.
https://doi.org/10.1073/pnas.211053698 |
| 34 | Parrinello S, Coppe JP, Krtolica A, Campisi J: Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci 2005;118:485-496.
https://doi.org/10.1242/jcs.01635 |
| 35 | Liu J, Xu K, Chase M, Ji Y, Logan JK, Buchsbaum RJ: Tiam1-regulated osteopontin in senescent fibroblasts contributes to the migration and invasion of associated epithelial cells. J Cell Sci 2012;125:376-386.
https://doi.org/10.1242/jcs.089466 |
| 36 | Yang G, Rosen DG, Zhang Z, Bast RC Jr, Mills GB, Colacino JA, Mercado-Uribe I, Liu J: The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A 2006;103:16472-16477.
https://doi.org/10.1073/pnas.0605752103 |
| 37 | Prieto LI, Baker DJ: Cellular Senescence and the Immune System in Cancer. Gerontology 2019;65:505-512.
https://doi.org/10.1159/000500683 |
| 38 | Marin I, Boix O, Garcia-Garijo A, Sirois I, Caballe A, Zarzuela E, Ruano I, Attolini CS, Prats N, López-Domínguez JA, Kovatcheva M, Garralda E, Muñoz J, Caron E, Abad M, Gros A, Pietrocola F, Serrano M: Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov 2023;13:410-431.
https://doi.org/10.1158/2159-8290.CD-22-0523 |
| 39 | Ruscetti M, Morris JP 4th, Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho YJ, Fennell M, Li J, Norgard RJ, Wilkinson JE, Alonso-Curbelo D, Sridharan R, Heller DA, de Stanchina E, Stanger BZ, Sherr CJ, Lowe SW: Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020;181:424-441.
https://doi.org/10.1016/j.cell.2020.03.008 |
| 40 | Costa TDF, Zhuang T, Lorent J, Turco E, Olofsson H, Masia-Balague M, Zhao M, Rabieifar P, Robertson N, Kuiper R, Sjölund J, Spiess M, Hernández-Varas P, Rabenhorst U, Roswall P, Ma R, Gong X, Hartman J, Pietras K, Adams PD, Defilippi P, Strömblad S: PAK4 suppresses RELB to prevent senescence-like growth arrest in breast cancer. Nat Commun 2019;10:3589.
https://doi.org/10.1038/s41467-019-11510-4 |
| 41 | Costa TDF, Strömblad S: New control of the senescence barrier in breast cancer. Mol Cell Oncol 2020;7:1684129.
https://doi.org/10.1080/23723556.2019.1684129 |
| 42 | Villot R, Poirier A, Bakan I, Boulay K, Fernández E, Devillers R, Gama-Braga L, Tribouillard L, Gagné A, Duchesne É, Caron D, Bérubé JS, Bérubé JC, Coulombe Y, Orain M, Gélinas Y, Gobeil S, Bossé Y, Masson JY, Elowe S, Bilodeau S, Manem V, Joubert P, Mallette FA, Laplante M: ZNF768 links oncogenic RAS to cellular senescence. Nat Commun 2021;12:4841.
https://doi.org/10.1038/s41467-021-24932-w |
| 43 | Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW: Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A 2007;104:13028-13033.
https://doi.org/10.1073/pnas.0701953104 |
| 44 | An B, Ji X, Gong Y: Role of CITED2 in stem cells and cancer. Oncol Lett 2020;20:107.
https://doi.org/10.3892/ol.2020.11968 |
| 45 | Chou YT, Hsieh CH, Chiou SH, Hsu CF, Kao YR, Lee CC, Chung CH, Wang YH, Hsu HS, Pang ST, Shieh YS, Wu CW: CITED2 functions as a molecular switch of cytokine-induced proliferation and quiescence. Cell Death Differ 2012;19:2015-2028.
https://doi.org/10.1038/cdd.2012.91 |
| 46 | Wang Z, Li Y, Wu D, Yu S, Wang Y, Leung Chan F: Nuclear receptor HNF4α performs a tumor suppressor function in prostate cancer via its induction of p21-driven cellular senescence. Oncogene 2020;39:1572-1589.
https://doi.org/10.1038/s41388-019-1080-3 |
| 47 | Donninger H, Calvisi DF, Barnoud T, Clark J, Schmidt ML, Vos MD, Clark GJ: NORE1A is a Ras senescence effector that controls the apoptotic/senescent balance of p53 via HIPK2.J Cell Biol 2015;208:777-789.
https://doi.org/10.1083/jcb.201408087 |
| 48 | Thaler S, Schmidt M, Schad A, Sleeman JP: RASSF1A inhibits estrogen receptor alpha expression and estrogen-independent signalling: implications for breast cancer development. Oncogene 2012;31:4912-22.
https://doi.org/10.1038/onc.2011.658 |
| 49 | Moulana FI, Priyani AAH, de Silva MVC, Dassanayake RS: BRAF-Oncogene-Induced Senescence and the Role of Thyroid-Stimulating Hormone Signaling in the Progression of Papillary Thyroid Carcinoma. Horm Cancer 2018;9:1-11.
https://doi.org/10.1007/s12672-017-0315-4 |
| 50 | Zou M, Baitei EY, Al-Rijjal RA, Parhar RS, Al-Mohanna FA, Kimura S, Pritchard C, Binessa HA, Alzahrani AS, Al-Khalaf HH, Hawwari A, Akhtar M, Assiri AM, Meyer BF, Shi Y: TSH overcomes Braf(V600E)-induced senescence to promote tumor progression via downregulation of p53 expression in papillary thyroid cancer. Oncogene 2016;35:1909-1918.
https://doi.org/10.1038/onc.2015.253 |
| 51 | Yang X, Qu K, Tao J, Yin G, Han S, Liu Q, Sun H: Inhibition of CIP2A attenuates tumor progression by inducing cell cycle arrest and promoting cellular senescence in hepatocellular carcinoma. Biochem Biophys Res Commun 2018;495:1807-1814.
https://doi.org/10.1016/j.bbrc.2017.11.124 |
| 52 | Diep CH, Charles NJ, Gilks CB, Kalloger SE, Argenta PA, Lange CA: Progesterone receptors induce FOXO1-dependent senescence in ovarian cancer cells. Cell Cycle 2013;12:1433-1449.
https://doi.org/10.4161/cc.24550 |
| 53 | Xu X, Liu Q, Zhang C, Ren S, Xu L, Zhao Z, Dou H, Li P, Zhang X, Gong Y, Shao C: Inhibition of DYRK1A-EGFR axis by p53-MDM2 cascade mediates the induction of cellular senescence. Cell Death Dis 2019;10:282.
https://doi.org/10.1038/s41419-019-1521-5 |
| 54 | Li Y, Fu M, Guo L, Sun X, Chen Y, Zhang T, Qin J, Shao X: Carboplatin-induced Senescence in Ovarian Cancer Cells Is Reversed Through EGFR and NF-κB. Signaling 2021, PREPRINT (Version 1) available at Research Square [https://doi.org/10.21203/rs.3.rs-215681/v1].
https://doi.org/10.21203/rs.3.rs-215681/v1 |
| 55 | Zhang M, Cai S, Zuo B, Gong W, Tang Z, Zhou D, Weng M, Qin Y, Wang S, Liu J, Ma F, Quan Z: Arctigenin induced gallbladder cancer senescence through modulating epidermal growth factor receptor pathway. Tumour Biol 2017;39:1010428317698359.
https://doi.org/10.1177/1010428317698359 |
| 56 | Senturk S, Mumcuoglu M, Gursoy-Yuzugullu O, Cingoz B, Akcali KC, Ozturk M: Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010;52:966-974.
https://doi.org/10.1002/hep.23769 |
| 57 | Jia Q, Xie B, Zhao Z, Huang L, Wei G, Ni T: Lung cancer cells expressing a shortened CDK16 3'UTR escape senescence through impaired miR-485-5p targeting. Mol Oncol 2022;16:1347-1364.
https://doi.org/10.1002/1878-0261.13125 |
| 58 | Pandey K, Park N, Park KS, Hur J, Cho YB, Kang M, An HJ, Kim S, Hwang S, Moon YW: Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers (Basel) 2020;12:3566.
https://doi.org/10.3390/cancers12123566 |
| 59 | Lei Q, Xia J, Feng X, Guo J, Li G, Zhou W. NEK2 promotes the progression of liver cancer by resisting the cellular senescence. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2022;47:153-164.
|
| 60 | He Q, Xue S, Tan Y, Zhang L, Shao Q, Xing L, Li Y, Xiang T, Luo X, Ren G: Dual inhibition of Akt and ERK signaling induces cell senescence in triple-negative breast cancer. Cancer Lett 2019;448:94-104.
https://doi.org/10.1016/j.canlet.2019.02.004 |
| 61 | Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, Marayati R, Kher S, George SD, Xu M, Wang-Gillam A, Samatar AA, Maitra A, Wennerberg K, Petricoin EF 3rd, Yin HH, Nelkin B, Cox AD, Yeh JJ, Der CJ: Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 2016;29:75-89.
https://doi.org/10.1016/j.ccell.2015.11.011 |
| 62 | Willobee BA, Gaidarski AA, Dosch AR, Castellanos JA, Dai X, Mehra S, Messaggio F, Srinivasan S, VanSaun MN, Nagathihalli NS, Merchant NB: Combined Blockade of MEK and CDK4/6 Pathways Induces Senescence to Improve Survival in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther 2021;20:1246-1256.
https://doi.org/10.1158/1535-7163.MCT-19-1043 |
| 63 | Zheng ZY, Chu MY, Lin W, Zheng YQ, Xu XE, Chen Y, Liao LD, Wu ZY, Wang SH, Li EM, Xu LY: Blocking STAT3 signaling augments MEK/ERK inhibitor efficacy in esophageal squamous cell carcinoma. Cell Death Dis 2022;13:496.
https://doi.org/10.1038/s41419-022-04941-3 |
| 64 | Jung SH, Hwang HJ, Kang D, Park HA, Lee HC, Jeong D, Lee K, Park HJ, Ko YG, Lee JS: mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2019;38:1639-1650.
https://doi.org/10.1038/s41388-018-0521-8 |
| 65 | Ke X, Li L, Li J, Zheng M, Liu P: Anti-oncogenic PTEN induces ovarian cancer cell senescence by targeting P21. Cell Biol Int 2022;46:118-128.
https://doi.org/10.1002/cbin.11709 |
| 66 | Sai X, Qin C, Wu Y, Zhao Y, Bian T: Downregulation of PTEN mediates bleomycin-induced premature senescence in lung cancer cells by suppressing autophagy. J Int Med Res 2020;48:300060520923522.
https://doi.org/10.1177/0300060520923522 |
| 67 | Kozlova NI, Morozevich GE, Berman AE: Implication of integrin α2β1 in senescence of SK-Mel-147 human melanoma cells. Aging (Albany NY) 2021;13:18006-18017.
https://doi.org/10.18632/aging.203309 |
| 68 | Ramachandran I, Ganapathy V, Gillies E, Fonseca I, Sureban SM, Houchen CW, Reis A, Queimado L: Wnt inhibitory factor 1 suppresses cancer stemness and induces cellular senescence. Cell Death Dis 2014;5:e1246.
https://doi.org/10.1038/cddis.2014.219 |
| 69 | Baskar R, Lee KA, Yeo R, Yeoh KW: Cancer and radiation therapy: current advances and future directions. Int J Med Sci 2012;9:193-199.
https://doi.org/10.7150/ijms.3635 |
| 70 | He X, Yang A, McDonald DG, Riemer EC, Vanek KN, Schulte BA, Wang GY: MiR-34a modulates ionizing radiation-induced senescence in lung cancer cells. Oncotarget 2017;8:69797-69807.
https://doi.org/10.18632/oncotarget.19267 |
| 71 | Jones KR, Elmore LW, Jackson-Cook C, Demasters G, Povirk LF, Holt SE, Gewirtz DA: p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol 2005;81:445-58.
https://doi.org/10.1080/09553000500168549 |
| 72 | Mirzayans R, Scott A, Cameron M, Murray D: Induction of accelerated senescence by gamma radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiat Res 2005;163:53-62.
https://doi.org/10.1667/RR3280 |
| 73 | Chen WS, Yu YC, Lee YJ, Chen JH, Hsu HY, Chiu SJ: Depletion of securin induces senescence after irradiation and enhances radiosensitivity in human cancer cells regardless of functional p53 expression. Int J Radiat Oncol Biol Phys 2010;77:566-574.
https://doi.org/10.1016/j.ijrobp.2009.12.013 |
| 74 | Yu X, Liu Y, Yin L, Peng Y, Peng Y, Gao Y, Yuan B, Zhu Q, Cao T, Xie B, Sun L, Chen Y, Gong Z, Qiu Y, Fan X, Li X: Radiation-promoted CDC6 protein stability contributes to radioresistance by regulating senescence and epithelial to mesenchymal transition. Oncogene 2019;38:549-563.
https://doi.org/10.1038/s41388-018-0460-4 |
| 75 | Schoetz U, Klein D, Hess J, Shnayien S, Spoerl S, Orth M, Mutlu S, Hennel R, Sieber A, Ganswindt U, Luka B, Thomsen AR, Unger K, Jendrossek V, Zitzelsberger H, Blüthgen N, Belka C, Unkel S, Klinger B, Lauber K: Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death Dis 2021;12:1162.
https://doi.org/10.1038/s41419-021-04454-5 |
| 76 | Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC, Hornyak TJ: EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res 2011;9:418-429.
https://doi.org/10.1158/1541-7786.MCR-10-0511 |
| 77 | Yu Y, Qi J, Xiong J, Jiang L, Cui D, He J, Chen P, Li L, Wu C, Ma T, Shao S, Wang J, Yu D, Zhou B, Huang D, Schmitt CA, Tao R: Epigenetic Co-Deregulation of EZH2/TET1 is a Senescence-Countering, Actionable Vulnerability in Triple-Negative Breast Cancer. Theranostics 2019;9:761-777.
https://doi.org/10.7150/thno.29520 |
| 78 | Filipczak PT, Leng S, Tellez CS, Do KC, Grimes MJ, Thomas CL, Walton-Filipczak SR, Picchi MA, Belinsky SA: p53-Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res 2019;79:1758-1768.
https://doi.org/10.1158/0008-5472.CAN-18-1234 |
| 79 | Chu L, Qu Y, An Y, Hou L, Li J, Li W, Fan G, Song BL, Li E, Zhang L, Qi W: Induction of senescence-associated secretory phenotype underlies the therapeutic efficacy of PRC2 inhibition in cancer. Cell Death Dis 2022;13:155.
https://doi.org/10.1038/s41419-022-04601-6 |
| 80 | Balakrishnan I, Danis E, Pierce A, Madhavan K, Wang D, Dahl N, Sanford B, Birks DK, Davidson N, Metselaar DS, Meel MH, Lemma R, Donson A, Vijmasi T, Katagi H, Sola I, Fosmire S, Alimova I, Steiner J, Gilani A, Hulleman E, Serkova NJ, Hashizume R, Hawkins C, Carcaboso AM, Gupta N, Monje M, Jabado N, Jones K, Foreman N, Green A, Vibhakar R, Venkataraman S: Senescence Induced by BMI1 Inhibition Is a Therapeutic Vulnerability in H3K27M-Mutant DIPG. Cell Rep 2020;33:108286.
https://doi.org/10.1016/j.celrep.2020.108286 |
| 81 | Tajima K, Matsuda S, Yae T, Drapkin BJ, Morris R, Boukhali M, Niederhoffer K, Comaills V, Dubash T, Nieman L, Guo H, Magnus NKC, Dyson N, Shioda T, Haas W, Haber DA, Maheswaran S: SETD1A protects from senescence through regulation of the mitotic gene expression program. Nat Commun 2019;10:2854.
https://doi.org/10.1038/s41467-019-10786-w |
| 82 | Ohta K, Haraguchi N, Kano Y, Kagawa Y, Konno M, Nishikawa S, Hamabe A, Hasegawa S, Ogawa H, Fukusumi T, Uemura M, Nishimura J, Hata T, Takemasa I, Mizushima T, Noguchi Y, Ozaki M, Kudo T, Sakai D, Satoh T, Fukami M, Ishii M, Yamamoto H, Doki Y, Mori M, Ishii H: Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int J Oncol 2013;42:1212-1218.
https://doi.org/10.3892/ijo.2013.1799 |
| 83 | Voelter-Mahlknecht S, Mahlknecht U: The sirtuins in the pathogenesis of cancer. Clin Epigenetics 2010;1:71-83.
https://doi.org/10.1007/s13148-010-0008-0 |
| 84 | Huang SB, Rivas P, Yang X, Lai Z, Chen Y, Schadler KL, Hu M, Reddick RL, Ghosh R, Kumar AP: SIRT1 inhibition-induced senescence as a strategy to prevent prostate cancer progression. Mol Carcinog 2022;61:702-716.
https://doi.org/10.1002/mc.23412 |
| 85 | Anwar T, Khosla S, Ramakrishna G: Increased expression of SIRT2 is a novel marker of cellular senescence and is dependent on wild type p53 status. Cell Cycle 2016;15:1883-1897.
https://doi.org/10.1080/15384101.2016.1189041 |
| 86 | George J, Nihal M, Singh CK, Zhong W, Liu X, Ahmad N: Pro-Proliferative Function of Mitochondrial Sirtuin Deacetylase SIRT3 in Human Melanoma. J Invest Dermatol;136:809-818.
https://doi.org/10.1016/j.jid.2015.12.026 |
| 87 | Huang FY, Wong DK, Seto WK, Mak LY, Cheung TT, Yuen MF: Tumor suppressive role of mitochondrial sirtuin 4 in induction of G2/M cell cycle arrest and apoptosis in hepatitis B virus-related hepatocellular carcinoma. Cell Death Discov 2021;7:88.
https://doi.org/10.1038/s41420-021-00470-8 |
| 88 | Lee N, Ryu HG, Kwon JH, Kim DK, Kim SR, Wang HJ, Kim KT, Choi KY: SIRT6 Depletion Suppresses Tumor Growth by Promoting Cellular Senescence Induced by DNA Damage in HCC. PLoS One 2016;11:e0165835.
https://doi.org/10.1371/journal.pone.0165835 |
| 89 | Ramzan F, Vickers MH, Mithen RF: Epigenetics, microRNA and Metabolic Syndrome: A Comprehensive Review. Int J Mol Sci 2021;22:5047.
https://doi.org/10.3390/ijms22095047 |
| 90 | Chen J, Wang M, Guo M, Xie Y, Cong YS: miR-127 regulates cell proliferation and senescence by targeting BCL6. PLoS One 2013;8:e80266.
https://doi.org/10.1371/journal.pone.0080266 |
| 91 | Ren XS, Yin MH, Zhang X, Wang Z, Feng SP, Wang GX, Luo YJ, Liang PZ, Yang XQ, He JX, Zhang BL: Tumor-suppressive microRNA-449a induces growth arrest and senescence by targeting E2F3 in human lung cancer cells. Cancer Lett 2014;344:195-203.
https://doi.org/10.1016/j.canlet.2013.10.031 |
| 92 | Wang J, Zheng X, Qin Z, Wei L, Lu Y, Peng Q, Gao Y, Zhang X, Zhang X, Li Z, Fu Y, Liu P, Liu C, Yan Q, Xiong W, Li G, Lu J, Ma J. Epstein-Barr virus miR-BART3-3p promotes tumorigenesis by regulating the senescence pathway in gastric cancer: J Biol Chem 2019;294:4854-4866.
https://doi.org/10.1074/jbc.RA118.006853 |
| 93 | Kanagasabai T, Li G, Shen TH, Gladoun N, Castillo-Martin M, Celada SI, Xie Y, Brown LK, Mark ZA, Ochieng J, Ballard BR, Cordon-Cardo C, Adunyah SE, Jin R, Matusik RJ, Chen Z: MicroRNA-21 deficiency suppresses prostate cancer progression through downregulation of the IRS1-SREBP-1 signaling pathway. Cancer Lett 2022;525:46-54.
https://doi.org/10.1016/j.canlet.2021.09.041 |
| 94 | Neault M, Mallette FA, Richard S: miR-137 Modulates a Tumor Suppressor Network-Inducing Senescence in Pancreatic Cancer Cells. Cell Rep 2016;14:1966-1978.
https://doi.org/10.1016/j.celrep.2016.01.068 |
| 95 | Weiner-Gorzel K, Dempsey E, Milewska M, McGoldrick A, Toh V, Walsh A, Lindsay S, Gubbins L, Cannon A, Sharpe D, O'Sullivan J, Murphy M, Madden SF, Kell M, McCann A, Furlong F: Overexpression of the microRNA miR-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med 2015;4:745-758.
https://doi.org/10.1002/cam4.409 |
| 96 | Weng JH, Yu CC, Lee YC, Lin CW, Chang WW, Kuo YL: miR-494-3p Induces Cellular Senescence and Enhances Radiosensitivity in Human Oral Squamous Carcinoma Cells. Int J Mol Sci 2016;17:1092.
https://doi.org/10.3390/ijms17071092 |
| 97 | Xu D, Takeshita F, Hino Y, Fukunaga S, Kudo Y, Tamaki A, Matsunaga J, Takahashi RU, Takata T, Shimamoto A, Ochiya T, Tahara H: miR-22 represses cancer progression by inducing cellular senescence. J Cell Biol 2011;193:409-424.
https://doi.org/10.1083/jcb.201010100 |
| 98 | Wang M, Cai WR, Meng R, Chi JR, Li YR, Chen AX, Yu Y, Cao XC: miR-485-5p suppresses breast cancer progression and chemosensitivity by targeting survivin. Biochem Biophys Res Commun 2018;501:48-54.
https://doi.org/10.1016/j.bbrc.2018.04.129 |
| 99 | Ye ZQ, Chen HB, Zhang TY, Chen Z, Tian L, Gu DN: MicroRNA-7 modulates cellular senescence to relieve gemcitabine resistance by targeting PARP1/NF-κB signaling in pancreatic cancer cells. Oncol Lett 2021;21:139.
https://doi.org/10.3892/ol.2020.12400 |
| 100 | Tazawa H, Tsuchiya N, Izumiya M, Nakagama H: Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A 2007;104:15472-15477.
https://doi.org/10.1073/pnas.0707351104 |
| 101 | Ramalho-Carvalho J, Graça I, Gomez A, Oliveira J, Henrique R, Esteller M, Jerónimo C: Downregulation of miR-130b~301b cluster is mediated by aberrant promoter methylation and impairs cellular senescence in prostate cancer. J Hematol Oncol 2017;10:43.
https://doi.org/10.1186/s13045-017-0415-1 |
| 102 | Liang Y, Yang W, Zhu Y, Yuan Y: Prognostic role of microRNA-203 in various carcinomas: evidence from a meta-analysis involving 13 studies. Springerplus 2016;5:1538.
https://doi.org/10.1186/s40064-016-3225-y |
| 103 | Noguchi S, Mori T, Otsuka Y, Yamada N, Yasui Y, Iwasaki J, Kumazaki M, Maruo K, Akao Y: Anti-oncogenic microRNA-203 induces senescence by targeting E2F3 protein in human melanoma cells. J Biol Chem 2012;287:11769-11777.
https://doi.org/10.1074/jbc.M111.325027 |
| 104 | Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, George J, Danson S, Kirkland JL: Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers (Basel) 2020;12:2134.
https://doi.org/10.3390/cancers12082134 |
| 105 | Lau L, Porciuncula A, Yu A, Iwakura Y, David G: Uncoupling the Senescence-Associated Secretory Phenotype from Cell Cycle Exit via Interleukin-1 Inactivation Unveils Its Protumorigenic Role. Mol Cell Biol 2019;39:e00586-e00618.
https://doi.org/10.1128/MCB.00586-18 |
| 106 | Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d'Adda di Fagagna F, Bernard D, Hernando E, Gil J: Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008;133:1006-1018.
https://doi.org/10.1016/j.cell.2008.03.038 |
| 107 | Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS: Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008;133:1019-1031.
https://doi.org/10.1016/j.cell.2008.03.039 |
| 108 | Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, Lowe SW: Non-cell-autonomous tumor suppression by p53 Cell 2013;153:449-460.
https://doi.org/10.1016/j.cell.2013.03.020 |
| 109 | Wang G, Cheng X, Zhang J, Liao Y, Jia Y, Qing C: Possibility of inducing tumor cell senescence during therapy. Oncol Lett 2021;22:496.
https://doi.org/10.3892/ol.2021.12757 |
| 110 | Xue W, Zender L, Miething C, Dickins RA, Hernando, E, Krizhanovsky V, Cordon-Cardo C, Lowe SW: Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656-660.
https://doi.org/10.1038/nature05529 |
| 111 | Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L: Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011;479:547-551.
https://doi.org/10.1038/nature10599 |
| 112 | Iannello A, Thompson TW, Ardolino M, Lowe SW, and Raulet DH: p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med 2013; 210:2057-2069.
https://doi.org/10.1084/jem.20130783 |
| 113 | Coppé JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J: Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6:e301
https://doi.org/10.1371/journal.pbio.0060301 |
| 114 | Wang T, Notta F, Navab R, Joseph J, Ibrahimov E, Xu J, Zhu CQ, Borgida A, Gallinger S, Tsao MS: Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol Cancer Res 2017;15:3-14.
https://doi.org/10.1158/1541-7786.MCR-16-0192 |
| 115 | Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, Egia A, Carver B, Gerald W, Cordon-Cardo C, Pandolfi PP: Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev 2008;22:2172-2177.
https://doi.org/10.1101/gad.1699608 |
| 116 | Ruhland MK, Loza AJ, Capietto AH: Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun 2016;7:11762.
https://doi.org/10.1038/ncomms11762 |
| 117 | Di GH, Liu Y, Lu Y, Liu J, Wu C, Duan HF: IL-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS One 2014;9:e113572.
https://doi.org/10.1371/journal.pone.0113572 |
| 118 | Zacarias-Fluck MF, Morancho B, Vicario R, Luque Garcia A, Escorihuela M, Villanueva J, Rubio IT, Arribas J: Effect of cellular senescence on the growth of HER2-positive breast cancers. J Natl Cancer Inst 2015;107:djv020.
https://doi.org/10.1093/jnci/djv020 |
| 119 | Liu J, Liu Y, Chen J, Hu C, Teng M, Jiao K, Shen Z, Zhu D, Yue J, Li Z, Li Y: The ROS-mediated activation of IL-6/STAT3 signaling pathway is involved in the 27-hydroxycholesterol-induced cellular senescence in nerve cells. Toxicol In vitro 2017;45:10-18.
https://doi.org/10.1016/j.tiv.2017.07.013 |
| 120 | Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D'Angelo R, Paulson AK, Chung S, Luther T, Paholak HJ, Liu S, Hassan KA, Zen Q, Clouthier SG, Wicha MS: Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 2012;47:570-584.
https://doi.org/10.1016/j.molcel.2012.06.014 |
| 121 | Li L, Huang Y, Gao Y, Shi T, Xu Y, Li H, Hyytiäinen M, Keski-Oja J, Jiang Q, Hu Y, Du Z: EGF/EGFR upregulates and cooperates with Netrin-4 to protect glioblastoma cells from DNA damage-induced senescence. BMC Cancer 2018;18:1215.
https://doi.org/10.1186/s12885-018-5056-4 |
| 122 | Sun X, Shi B, Zheng H, Min L, Yang J, Li X, Liao X, Huang W, Zhang M, Xu S, Zhu Z, Cui H, Liu X: Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis 2018;9:260.
https://doi.org/10.1038/s41419-018-0303-9 |
| 123 | Romaniello D, Gelfo V, Pagano F, Ferlizza E, Sgarzi M, Mazzeschi M, Morselli A, Miano C, D'Uva G, Lauriola M: Senescence-associated reprogramming induced by interleukin-1 impairs response to EGFR neutralization. Cell Mol Biol Lett 2022;27:20.
https://doi.org/10.1186/s11658-022-00319-7 |
| 124 | Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, Heikenwalder M, Wang XW, Zender L, Greten TF: Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016;30:533-547.
https://doi.org/10.1016/j.ccell.2016.09.003 |
| 125 | Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, Mendoza-Parra MA, Kanashova T, Metzner M, Pardon K, Reimann M, Trumpp A, Dörken B, Zuber J, Gronemeyer H, Hummel M, Dittmar G, Lee S, Schmitt CA: Senescence-associated reprogramming promotes cancer stemness. Nature 2018;553:96-100.
https://doi.org/10.1038/nature25167 |
| 126 | Basu SK, Lee S, Salotti J, Basu S, Sakchaisri K, Xiao Z, Walia V, Westlake CJ, Morrison DK, Johnson PF: Oncogenic RAS-Induced Perinuclear Signaling Complexes Requiring KSR1 Regulate Signal Transmission to Downstream Targets. Cancer Res 2018;78:891-908.
https://doi.org/10.1158/0008-5472.CAN-17-2353 |
| 127 | Nacarelli T, Fukumoto T, Zundell JA, Fatkhutdinov N, Jean S, Cadungog MG, Borowsky ME, Zhang R: NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res 2020;80:890-900.
https://doi.org/10.1158/0008-5472.CAN-19-2830 |
| 128 | Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM: The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 2017;31:172-183.
https://doi.org/10.1101/gad.290635.116 |
| 129 | Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Muñoz-Martin M, Blanco-Aparicio C, Pastor J, Gómez-López G, De Martino A, Blasco MA, Abad M, Serrano M: Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016;354:aaf4445.
https://doi.org/10.1126/science.aaf4445 |
| 130 | Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, Vallier L, Gil J: Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev 2009;23:2134-2139.
https://doi.org/10.1101/gad.1811609 |
| 131 | Xu H, Lin F, Wang Z, Yang L, Meng J, Ou Z, Shao Z, Di G, Yang G: CXCR2 promotes breast cancer metastasis and chemoresistance via suppression of AKT1 and activation of COX2. Cancer Lett 2018;412:69-80.
https://doi.org/10.1016/j.canlet.2017.09.030 |
| 132 | Medema JP: Escape from senescence boosts tumour growth. Nature 2018;553:37-38.
https://doi.org/10.1038/d41586-017-08652-0 |
| 133 | Wang A, Jiang A, Gan X, Wang Z, Huang J, Dong K, Liu B, Wang L, Chen M: EGFR-AS1 Promotes Bladder Cancer Progression by Upregulating EGFR. Biomed Res Int 2020;2020:6665974.
https://doi.org/10.1155/2020/6665974 |
| 134 | Ahmad I, Patel R, Singh LB, Nixon C, Seywright M, Barnetson RJ, Brunton VG, Muller WJ, Edwards J, Sansom OJ, Leung HY: HER2 overcomes PTEN (loss)-induced senescence to cause aggressive prostate cancer. Proc Natl Acad Sci U S A 2011;108:16392-16397.
https://doi.org/10.1073/pnas.1101263108 |
| 135 | Parisotto M, Grelet E, El Bizri R, Metzger D: Senescence controls prostatic neoplasia driven by Pten loss. Mol Cell Oncol 2018;6:1511205.
https://doi.org/10.1080/23723556.2018.1511205 |
| 136 | Liu Y, Yang S, Wang K, Lu J, Bao X, Wang R, Qiu Y, Wang T, Yu H: Cellular senescence and cancer: Focusing on traditional Chinese medicine and natural products. Cell Prolif 2020;53:e12894.
https://doi.org/10.1111/cpr.12894 |
| 137 | Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, Altman RB: Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics 2011;21:440-446.
https://doi.org/10.1097/FPC.0b013e32833ffb56 |
| 138 | Yang MY, Lin PM, Liu YC, Hsiao HH, Yang WC, Hsu JF, Hsu CM, Lin SF: Induction of cellular senescence by doxorubicin is associated with upregulated miR-375 and induction of autophagy in K562 cells. PLoS One 2012;7:e37205.
https://doi.org/10.1371/journal.pone.0037205 |
| 139 | Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB: A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res 1999;59:3761-3767.
|
| 140 | Wang X, Wong SC, Pan J, Tsao SW, Fung KH, Kwong DL, Sham JS, Nicholls JM: Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res 1998;58:5019-5022.
|
| 141 | Günther W, Pawlak E, Damasceno R, Arnold H, Terzis AJ: Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br J Cancer 2003;88:463-469.
https://doi.org/10.1038/sj.bjc.6600711 |
| 142 | Hirose Y, Katayama M, Mirzoeva OK, Berger MS, Pieper RO: Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res 2005;65:4861-4869.
https://doi.org/10.1158/0008-5472.CAN-04-2633 |
| 143 | Ohba S, Hirose Y, Kawase T, Sano H: Inhibition of c-Jun N-terminal kinase enhances temozolomide-induced cytotoxicity in human glioma cells. J Neurooncol 2009;95:307-316.
https://doi.org/10.1007/s11060-009-9929-x |
| 144 | Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, Kaina B: Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One 2013;8:e55665.
https://doi.org/10.1371/journal.pone.0055665 |
| 145 | Mhaidat NM, Zhang XD, Allen J, Avery-Kiejda KA, Scott RJ, Hersey P: Temozolomide induces senescence but not apoptosis in human melanoma cells. Br J Cancer 2007;97:1225-1233.
https://doi.org/10.1038/sj.bjc.6604017 |
| 146 | Jaiswal AS, Panda H, Law BK, Sharma J, Jani J, Hromas R, Narayan S: NSC666715 and Its Analogs Inhibit Strand-Displacement Activity of DNA Polymerase β and Potentiate Temozolomide-Induced DNA Damage, Senescence and Apoptosis in Colorectal Cancer Cells. PLoS One 2015;10:e0123808.
https://doi.org/10.1371/journal.pone.0123808 |
| 147 | Mikuła-Pietrasik J, Witucka A, Pakuła M, Uruski P, Begier-Krasińska B, Niklas A, Tykarski A, Książek K: Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell Mol Life Sci 2019;76:681-697.
https://doi.org/10.1007/s00018-018-2954-1 |
| 148 | Hu W, Sung T, Jessen BA, Thibault S, Finkelstein MB, Khan NK, Sacaan AI: Mechanistic Investigation of Bone Marrow Suppression Associated with Palbociclib and its Differentiation from Cytotoxic Chemotherapies. Clin Cancer Res 2016;22:2000-8.
https://doi.org/10.1158/1078-0432.CCR-15-1421 |
| 149 | Scheule RK, Perkins RC, Hamilton R, Holian A: Bleomycin stimulation of cytokine secretion by the human alveolar macrophage. Am J Physiol 1992;262:L386-391.
https://doi.org/10.1152/ajplung.1992.262.4.L386 |
| 150 | Qiu T, Tian Y, Gao Y, Ma M, Li H, Liu X, Wu H, Zhang Y, Ding H, Cao M, Zhang J, Dai J, Chen J, Cai H: PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging (Albany NY) 2019;11:7492-7509.
https://doi.org/10.18632/aging.102262 |
| 151 | Linge A, Weinhold K, Bläsche R, Kasper M, Barth K: Downregulation of caveolin-1 affects bleomycin-induced growth arrest and cellular senescence in A549 cells. Int J Biochem Cell Biol 2007;39:1964-1974.
https://doi.org/10.1016/j.biocel.2007.05.018 |
| 152 | Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen J, Cai H: Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell 2019;18:e12858.
https://doi.org/10.1111/acel.12858 |
| 153 | DeMasters GA, Gupta MS, Jones KR, Cabot M, Wang H, Gennings C, Park M, Bratland A, Ree AH, Gewirtz DA: Potentiation of cell killing by fractionated radiation and suppression of proliferative recovery in MCF-7 breast tumor cells by the Vitamin D3 analog EB 1089. J Steroid Biochem Mol Biol 2004;92:365-374.
https://doi.org/10.1016/j.jsbmb.2004.07.011 |
| 154 | Jones KR, Elmore LW, Jackson-Cook C, Demasters G, Povirk LF, Holt SE, Gewirtz DA: p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol 2005;81:445-458.
https://doi.org/10.1080/09553000500168549 |
| 155 | Quick QA, Gewirtz DA: An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J Neurosurg 2006;105:111-118.
https://doi.org/10.3171/jns.2006.105.1.111 |
| 156 | Malumbres M, Barbacid M: Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153-166.
https://doi.org/10.1038/nrc2602 |
| 157 | Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, Li Y, Carpenter EL, Attiyeh EF, Diskin SJ, Kim S, Parasuraman S, Caponigro G, Schnepp RW, Wood AC, Pawel B, Cole KA, Maris JM: Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res 2013;19:6173-6182.
https://doi.org/10.1158/1078-0432.CCR-13-1675 |
| 158 | Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL: Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004;3:1427-1438.
https://doi.org/10.1158/1535-7163.1427.3.11 |
| 159 | O'Leary B, Finn RS, Turner NC: Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 2016 ;13:417-430.
https://doi.org/10.1038/nrclinonc.2016.26 |
| 160 | Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, Raghavendra AS, Zhao Y, Bashour SI, Ibrahim NK, Karuturi M, Wang J, Winkler JD, Amaravadi RK, Hunt KK, Tripathy D, Keyomarsi K: CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 2017;8:15916.
https://doi.org/10.1038/ncomms15916 |
| 161 | Leontieva OV, Gudkov AV, Blagosklonny MV: Weak p53 permits senescence during cell cycle arrest. Cell Cycle 2010;9:4323-7.
https://doi.org/10.4161/cc.9.21.13584 |
| 162 | Yoshida A, Lee EK, Diehl JA: Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res 2016;76:2990-3002.
https://doi.org/10.1158/0008-5472.CAN-15-2931 |
| 163 | Muñoz-Espín D, Rovira M, Galiana I, Giménez C, Lozano-Torres B, Paez-Ribes M, Llanos S, Chaib S, Muñoz-Martín M, Ucero AC, Garaulet G, Mulero F, Dann SG, VanArsdale T, Shields DJ, Bernardos A, Murguía JR, Martínez-Máñez R, Serrano M: A versatile drug delivery system targeting senescent cells. EMBO Mol Med 2018;10:e9355.
https://doi.org/10.15252/emmm.201809355 |
| 164 | Yang MY, Lin PM, Liu YC, Hsiao HH, Yang WC, Hsu JF, Hsu CM, Lin SF: Induction of cellular senescence by doxorubicin is associated with upregulated miR-375 and induction of autophagy in K562 cells. PLoS One 2012;7:e37205.
https://doi.org/10.1371/journal.pone.0037205 |
| 165 | Kovatcheva M, Liu DD, Dickson MA, Klein ME, O'Connor R, Wilder FO, Socci ND, Tap WD, Schwartz GK, Singer S, Crago AM, Koff A: MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget 2015;6:8226-8243.
https://doi.org/10.18632/oncotarget.3364 |
| 166 | Kovatcheva M, Liao W, Klein ME, Robine N, Geiger H, Crago AM, Dickson MA, Tap WD, Singer S, Koff A: ATRX is a regulator of therapy induced senescence in human cells. Nat Commun 2017;8:386.
https://doi.org/10.1038/s41467-017-00540-5 |
| 167 | Klein ME, Dickson MA, Antonescu C, Qin LX, Dooley SJ, Barlas A, Manova K, Schwartz GK, Crago AM, Singer S, Koff A, Tap WD: PDLIM7 and CDH18 regulate the turnover of MDM2 during CDK4/6 inhibitor therapy-induced senescence. Oncogene 2018;37:5066-5078.
https://doi.org/10.1038/s41388-018-0332-y |
| 168 | Valenzuela CA, Vargas L, Martinez V, Bravo S, Brown NE: Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp Cell Res 2017;360:390-396.
https://doi.org/10.1016/j.yexcr.2017.09.031 |
| 169 | Bollard J, Miguela V, Ruiz de Galarreta M, Venkatesh A, Bian CB, Roberto MP, Tovar V, Sia D, Molina-Sánchez P, Nguyen CB, Nakagawa S, Llovet JM, Hoshida Y, Lujambio A: Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017;66:1286-1296.
https://doi.org/10.1136/gutjnl-2016-312268 |
| 170 | Guenther LM, Dharia NV, Ross L, Conway A, Robichaud AL, Catlett JL 2nd, Wechsler CS, Frank ES, Goodale A, Church AJ, Tseng YY, Guha R, McKnight CG, Janeway KA, Boehm JS, Mora J, Davis MI, Alexe G, Piccioni F, Stegmaier K: A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin Cancer Res 2019;25:1343-1357.
https://doi.org/10.1158/1078-0432.CCR-18-0372 |
| 171 | Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, Ramm S, Palmer AC, Yuzugullu H, Varadan V, Tuck D, Harris LN, Wong KK, Liu XS, Sicinski P, Winer EP, Krop IE, Zhao JJ: Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016;29:255-269.
https://doi.org/10.1016/j.ccell.2016.02.006 |
| 172 | Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, Hoog J, Ellis MJ, Ma CX, Ramm S, Krop IE, Winer EP, Roberts TM, Kim HJ, McAllister SS, Zhao JJ: CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017;548:471-475.
https://doi.org/10.1038/nature23465 |
| 173 | Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, Dowless M, Dempsey J, Zeng Y, Torres R, Boehnke K, Mur C, Marugán C, Baquero C, Yu C, Bray SM, Wulur IH, Bi C, Chu S, Qian HR, Iversen PW, Merzoug FF, Ye XS, Reinhard C, De Dios A, Du J, Caldwell CW, Lallena MJ, Beckmann RP, Buchanan SG: Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017;32:761-776.
https://doi.org/10.1016/j.ccell.2017.11.006 |
| 174 | Torres-Guzmán R, Calsina B, Hermoso A, Baquero C, Alvarez B, Amat J, McNulty AM, Gong X, Boehnke K, Du J, de Dios A, Beckmann RP, Buchanan S, Lallena MJ: Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017;8:69493-69507.
https://doi.org/10.18632/oncotarget.17778 |
| 175 | Schaer DA, Beckmann RP, Dempsey JA, Huber L, Forest A, Amaladas N, Li Y, Wang YC, Rasmussen ER, Chin D, Capen A, Carpenito C, Staschke KA, Chung LA, Litchfield LM, Merzoug FF, Gong X, Iversen PW, Buchanan S, de Dios A, Novosiadly RD, Kalos M: The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep 2018;22:2978-2994.
https://doi.org/10.1016/j.celrep.2018.02.053 |
| 176 | Amatori S, Bagaloni I, Viti D, Fanelli M: Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer 2011;71:113-115.
https://doi.org/10.1016/j.lungcan.2010.10.016 |
| 177 | Putri JF, Widodo N, Sakamoto K, Kaul SC, Wadhwa R: Induction of senescence in cancer cells by 5'-Aza-2'-deoxycytidine: Bioinformatics and experimental insights to its targets. Comput Biol Chem 2017;70:49-55.
https://doi.org/10.1016/j.compbiolchem.2017.08.003 |
| 178 | Venturelli S, Berger A, Weiland T, Essmann F, Waibel M, Nuebling T, Häcker S, Schenk M, Schulze-Osthoff K, Salih HR, Fulda S, Sipos B, Johnstone RW, Lauer UM, Bitzer M: Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2'-deoxycytidine in solid tumor cells. Mol Cancer Ther 2013;12:2226-36
https://doi.org/10.1158/1535-7163.MCT-13-0137 |
| 179 | Borghesan M, Fusilli C, Rappa F, Panebianco C, Rizzo G, Oben JA, Mazzoccoli G, Faulkes C, Pata I, Agodi A, Rezaee F, Minogue S, Warren A, Peterson A, Sedivy JM, Douet J, Buschbeck M, Cappello F, Mazza T, Pazienza V, Vinciguerra M: DNA Hypomethylation and Histone Variant macroH2A1 Synergistically Attenuate Chemotherapy-Induced Senescence to Promote Hepatocellular Carcinoma Progression. Cancer Res 2016;76:594-606.
https://doi.org/10.1158/0008-5472.CAN-15-1336 |
| 180 | Xu WS, Perez G, Ngo L, Gui CY, Marks PA: Induction of polyploidy by histone deacetylase inhibitor: a pathway for antitumor effects. Cancer Res 2005;65:7832-7839.
https://doi.org/10.1158/0008-5472.CAN-04-4608 |
| 181 | Elknerova K, Myslivcova D, Lacinova Z, Marinov I, Uherkova L, Stöckbauer P: Epigenetic modulation of gene expression of human leukemia cell lines - induction of cell death and senescence. Neoplasma 2011;58:35-44.
https://doi.org/10.4149/neo_2011_01_35 |
| 182 | Kaletsch A, Pinkerneil M, Hoffmann MJ, Jaguva Vasudevan AA, Wang C, Hansen FK, Wiek C, Hanenberg H, Gertzen C, Gohlke H, Kassack MU, Kurz T, Schulz WA, Niegisch G: Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin Epigenetics 2018;10:100.
https://doi.org/10.1186/s13148-018-0531-y |
| 183 | Däbritz JH, Yu Y, Milanovic M, Schönlein M, Rosenfeldt MT, Dörr JR, Kaufmann AM, Dörken B, Schmitt CA: CD20-Targeting Immunotherapy Promotes Cellular Senescence in B-Cell Lymphoma. Mol Cancer Ther 2016;15:1074-1081.
https://doi.org/10.1158/1535-7163.MCT-15-0627 |
| 184 | Shukla Y, Singh M: Cancer preventive properties of ginger: a brief review. Food Chem Toxicol 2007;45:683‐690.
https://doi.org/10.1016/j.fct.2006.11.002 |
| 185 | Kundu JK, Na HK, Surh YJ: Ginger‐derived phenolic substances with cancer preventive and therapeutic potential. Forum Nutr 2009;61:182‐192.
https://doi.org/10.1159/000212750 |
| 186 | Kaewtunjai N, Wongpoomchai R, Imsumran A, Pompimon W, Athipornchai A, Suksamrarn A, Lee TR, Tuntiwechapikul W. Ginger extract promotes telomere shortening and cellular senescence in A549 lung cancer cells. AcsOmega 2018;3:18572‐18581.
https://doi.org/10.1021/acsomega.8b02853 |
| 187 | McCormack D, McFadden D. Pterostilbene and cancer: current review. J Surg Res 2012;173:e53‐e61.
https://doi.org/10.1016/j.jss.2011.09.054 |
| 188 | Tippani R, Prakhya LJ, Porika M, Sirisha K, Abbagani S, Thammidala C: Pterostilbene as a potential novel telomerase inhibitor: molecular docking studies and its in vitro evaluation. Curr Pharm Biotechnol 2014;14:1027-1035.
https://doi.org/10.2174/1389201015666140113112820 |
| 189 | Chen RJ, Wu PH, Ho CT, Way TD, Pan MH, Chen HM, Ho YS, Wang YJ : P53‐dependent downregulation of hTERT protein expression and telomerase activity induces senescence in lung cancer cells as a result of pterostilbene treatment. Cell Death Dis 2017;8:e2985.
https://doi.org/10.1038/cddis.2017.333 |
| 190 | Liu H, Dong Y, Gao Y, Du Z, Wang Y, Cheng P, Chen A, Huang H: The fascinating effects of baicalein on cancer: a review. Int J Mol Sci 2016;17:e1681.
https://doi.org/10.3390/ijms17101681 |
| 191 | Dou J, Wang Z, Ma L, Peng B, Mao K, Li C, Su M, Zhou C, Peng G: Baicalein and baicalin inhibit colon cancer using two distinct fashions of apoptosis and senescence. Oncotarget 2018;9:20089‐20102.
https://doi.org/10.18632/oncotarget.24015 |
| 192 | Hou ZY, Tong XP, Peng YB, Zhang BK, Yan M: Broad targeting of triptolide to resistance and sensitization for cancer therapy. Biomed Pharmacother 2018;104:771‐780.
https://doi.org/10.1016/j.biopha.2018.05.088 |
| 193 | Li R, Zhang X, Tian X, Shen C, Zhang Q, Zhang Y, Wang Z, Wang F, Tao Y : Triptolide inhibits tumor growth by induction of cellular senescence. Oncol Rep 2017;37:442‐448.
https://doi.org/10.3892/or.2016.5258 |
| 194 | Ebrahimi SA, Saghaeian‐Shahri T, Shafiei M, Rostami P, Mahmoudian M: Interaction of papaverine with the enalapril‐induced cough in guinea pig. Acta Physiol Hung 2006;93:71‐78.
https://doi.org/10.1556/APhysiol.93.2006.1.8 |
| 195 | Noureini SK, Wink M: Antiproliferative effect of the isoquinoline alkaloid papaverine in hepatocarcinoma HepG‐2 cells-inhibition of telomerase and induction of senescence. Molecules 2014;19:11846‐11859.
https://doi.org/10.3390/molecules190811846 |
| 196 | Patel K, Gadewar M, Tripathi R, Prasad SK, Patel DK: A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta‐carboline alkaloid "Harmine". Asian Pac J Trop Biomed 2012;2:660‐664.
https://doi.org/10.1016/S2221-1691(12)60116-6 |
| 197 | Zhao L, Wink M: The beta‐carboline alkaloid harmine inhibits telomerase activity of MCF‐7 cells by down‐regulating hTERT mRNA expression accompanied by an accelerated senescent phenotype. PeerJ 2013;1:e174.
https://doi.org/10.7717/peerj.174 |
| 198 | Miller S, Stagl J, Wallerstedt DB, Ryan M, Mansky PJ: Botanicals used in complementary and alternative medicine treatment of cancer: clinical science and future perspectives. Expert Opin Inv Drug 2008;17:1353‐1364.
https://doi.org/10.1517/13543784.17.9.1353 |
| 199 | Lu S, Qin X, Yuan S, Li Y, Wang L, Jin Y, Zeng G, Yen L, Hu J, Dang T, Song S, Hou Q, Rao J: Effect of Tianshengyuan‐1 (TSY‐1) on telomerase activity and hematopoietic recovery ‐ In vitro, ex vivo, and in vivo studies. Int J Clin Exp Med 2014;7:597‐606.
|
| 200 | Yu W, Qin X, Jin Y, Santiskulvong C, Vu V, Zeng G, Zhang Z, Chow M, Rao J:. Tianshengyuan‐1 (TSY‐1) regulates cellular Telomerase activity by methylation of TERT promoter. Oncotarget 2017;8:7977‐7988.
https://doi.org/10.18632/oncotarget.13939 |
| 201 | Hankinson SE, Eliassen AH: Endogenous estrogen, testosterone and progesterone levels in relation to breast cancer risk. J Steroid Biochem Mol Biol 2007;106:24-30.
https://doi.org/10.1016/j.jsbmb.2007.05.012 |
| 202 | Bajbouj K, Shafarin J, Taneera J, Hamad M: Estrogen Signaling Induces Mitochondrial Dysfunction-Associated Autophagy and Senescence in Breast Cancer Cells. Biology (Basel) 2020;9:68.
https://doi.org/10.3390/biology9040068 |
| 203 | Atif F, Yousuf S, Espinosa-Garcia C, Sergeeva E, Stein DG: Progesterone Treatment Attenuates Glycolytic Metabolism and Induces Senescence in Glioblastoma. Sci Rep 2019;9:988.
https://doi.org/10.1038/s41598-018-37399-5 |
| 204 | Dai D, Wolf DM, Litman ES, White MJ, Leslie KK: Progesterone inhibits human endometrial cancer cell growth and invasiveness: down-regulation of cellular adhesion molecules through progesterone B receptors. Cancer Res 2002;62:881-886.
|
| 205 | Bajalovic N, Or YZ, Woo ARE, Lee SH, Lin VCL: High Levels of Progesterone Receptor B in MCF-7 Cells Enable Radical Anti-Tumoral and Anti-Estrogenic Effect of Progestin. Biomedicines 2022;10:1860.
https://doi.org/10.3390/biomedicines10081860 |
| 206 | Wang H, Shi H: Megestrol acetate drives endometrial carcinoma cell senescence via interacting with progesterone receptor B/FOXO1 axis. Exp Biol Med (Maywood) 2021;246:2307-2316.
https://doi.org/10.1177/15353702211026566 |
| 207 | Chang J, Wu Y, Zhou S, Tian Y, Wang Y, Tian J, Song W, Dong Y, Li J, Zhao Z, Che G: Genetically predicted testosterone and cancers risk in men: a two-sample Mendelian randomization study. J Transl Med 2022;20:573.
https://doi.org/10.1186/s12967-022-03783-z |
| 208 | Gravina GL, Marampon F, Sanità P, Festuccia C, Forcella C, Scarsella L, Jitariuc A, Vetuschi A, Sferra R, Colapietro A, Carosa E, Dolci S, Lenzi A, Jannini EA: Episode-like pulse testosterone supplementation induces tumor senescence and growth arrest down-modulating androgen receptor through modulation of p-ERK1/2, pARser81 and CDK1 signaling: biological implications for men treated with testosterone replacement therapy. Oncotarget 2017;8:113792-113806.
https://doi.org/10.18632/oncotarget.22776 |