Pancreas Β-Cells in Type 1 and Type 2 Diabetes: Cell Death, Oxidative Stress and Immune Regulation. Recently Appearing Changes in Diabetes Consequences
Keywords
Abstract
Introduction
Diabetes mellitus is a group of diseases characterized by high blood glucose levels (hyperglycemia) caused by the inability of pancreatic β-cells to secrete enough insulin. Type 1 diabetes (T1D), which accounts for approximately 5% of all cases of diabetes [1], occurs when β-cells are selectively destroyed by an autoimmune process. The onset of autoimmune diabetes is preceded by infiltration of pancreatic islets by immune cells. Ultimately, impaired tolerance to self-antigens allows autoreactive T cells to activate and attack β-cells, resulting in a loss of insulin secretion. However, innate immune cells such as macrophages and dendritic cells (DCs) are the first to enter the islets in insulitis [2-4]. Type 2 diabetes (T2D), which accounts for 90-95% of all cases [4], develops due to the inability of β-cells to produce enough insulin to meet the needs of a human with respect to aging, inactivity, obesity, or genetic risk factors. In addition, approximately 10% of T2D patients aged 35 years and older develop latent autoimmune diabetes, which requires treatment with exogenous insulin [5, 6]. Oxidative stress, which occurs when the balance between the production and removal of reactive oxygen species (ROS) is disturbed, can contribute to the damage of β-cells in both T1D and T2D [5]. ROS, such as superoxide and hydrogen peroxide, cause oxidation of lipids, proteins, and DNA, which can eventually lead to cell death. Indeed, numerous studies have reported increased oxidative damage to islets in rodents and diabetic patients [6, 7]. In addition, β-cells are considered vulnerable to oxidative stress due to reduced expression of antioxidants, including catalase and glutathione peroxidase, compared to their levels in other tissues [8]. Both forms of diabetes mellitus are characterized by abnormalities in pancreatic islet beta cells, leading to their death in T1D and accelerated apoptosis in T2D. The resulting chronic hyperglycemia leads to chronic oxidative stress in all tissues because glucose at abnormally high concentrations forms reactive oxygen species. It has been repeatedly emphasized that this can lead to oxidative damage of classic secondary targets of diabetes, such as the eyes, kidneys, nerves, and blood vessels [9]. It is less well known that the beta-cell itself is also a prime target, which multiplies the risk. This adverse effect of high glucose concentrations is called glucose toxicity. The main manifestation of glucose toxicity in β-cells is anomalous insulin gene expression, decreased insulin levels, and defective insulin secretion.
Published paper discussed the underlying mechanism by which resident (pancreatic) and circulating macrophages regulate β-cell development and survival in several scenarios, including T2D, T1D, obesity, oxidative stress, and insulin resistance [9]. Additionally, earlier studies have shown that the antioxidant enzymes peroxiredoxins are expressed in β-cells and protect them from various types of oxidative damage. The roles of peroxiredoxins and their partners, thioredoxin and thioredoxin reductase, in β-cell antioxidant defense are well known [10, 11]. We have previously demonstrated the protective role of exogenous peroxiredoxin 6 under T1D conditions in a diabetic mouse model and in a rat insulinoma RIN-m5F β-cell model [12-14]. In addition, we have recently discussed the role of senescent β-cells in the development of type 1 diabetes, in which we concluded that peroxiredoxin 6 can be qualified as a new protective and senolytic agent in diabetes mellitus [15].
The role of oxidative stress in T1D and T2D development
Oxidative stress, which occurs when the balance between the production and removal of reactive oxygen species (ROS) is disturbed, can contribute to damage to β-cells in both T1D and T2D [16]. ROS, such as superoxide and hydrogen peroxide, cause the oxidation of lipids, proteins, and DNA, which can ultimately lead to cell death [17]. Indeed, numerous studies have indicated increased oxidative damage to islets in rodents and diabetic patients [18, 19]. In addition, β-cells are considered to be particularly vulnerable to oxidative stress due to reduced expression of antioxidants, including catalase and glutathione peroxidase, compared to other tissues [8]. Oxidative stress occurs when ROS are produced at levels that overcome the antioxidant capacity of the cell. Recent studies have shown high expression of various isoforms of peroxiredoxins, thioredoxin and thioredoxin reductase in β-cells and provided experimental evidence supporting the role of these enzymes in promoting β-cell function and survival in response to various oxidative stressors [10, 11].
T1D, which accounts for approximately 5% of all cases of diabetes mellitus [20], occurs when β-cells are selectively destroyed by an autoimmune process. The onset of autoimmune diabetes is preceded by infiltration of pancreatic islets by immune cells. Innate immune cells such as macrophages and dendritic cells (DCs) are the first to enter islets in insulitis [20]. T2D, which accounts for 90-95% of all diabetic disease cases [1], develops due to the inability of β-cells to produce enough insulin to meet their needs with respect to aging, inactivity, obesity, or genetic risk factors. In addition, approximately 10% of T2D patients aged 35 years and older develop latent autoimmune diabetes, which requires treatment with exogenous insulin [21].
The endoplasmic reticulum (ER) is a center of protein synthesis, folding, modification, and transport, as well as a site of biosynthesis of phospholipids and cholesterol [22, 23]. Our current knowledge suggests that along with genetic defects, dysfunctions of organelles in β-cells contribute to the earliest stages of the disease as well as other factors, such as excessive production of reactive oxygen species and a decrease in volume and mass of beta-cells due to apoptosis [24]. Induction of cellular stress by external or internal players triggers several coordinated intracellular processes that may lead to either restoring cellular homeostasis or committing to cell death. Those include the response to unfolded proteins, autophagy induction, hypoxia response, and regulation of mitochondrial functions, all of these are parts of the global endoplasmic reticulum stress (ERS) response [25].
Like the ER, mitochondria are complex and dynamic cellular organelles that play a key role in β-cell functions, in particular by linking glucose metabolism to insulin secretion, as well as in the regulation of apoptotic cell death through the production of ROS and release of cytochrome C [26]. In addition, the formation of amyloid as well as stress-induced dysfunctions of β-cells and their apoptosis were shown to play important roles in the development of T1D [27-30]. However, the interplay between the ER and mitochondria in a mechanism of adaptation to environmental stress indicates that both organelles orchestrate the link between beta cells and the immune system.
Dysfunctional mitochondria have been studied in the context of metabolic disorders and T2D, where they have been linked for decades to insulin resistance and β-cell deficiency. Although β-cell deficiency in pancreatic islets differs in many aspects between T1D and T2D, the islet-specific inflammatory microenvironment that is characterized by elevated local levels of proinflammatory cytokines [31, 32], together with increased recruitment and activation of innate and adaptive immune cells (e.g., macrophages, B cells and T cells) [33-34] in tissues and accumulation of amyloid deposits [35], is a common feature of both pathologies. Metformin, GLP-1 analogs, SGLT-2 inhibitors, and the L-VGCC inhibitor verapamil, which either modulate mitochondrial bioenergetics/ROS (metformin), ER stress (GLP-1 analogs, [36, 37], toxic glucose (SGLT-2 inhibitors) [38] or cellular Ca2+ homeostasis (verapamil) [39], are currently being studied in clinical trials.
Another consequence of ER stress and mitochondrial dysfunction is the induction of cellular senescence [40] secondary to the increase in ROS production and redox balance impairment [41]. Cell aging (senescence) is a complex response that determines cell fate and is characterized by the formation of senescence-associated secretory phenotypes (SASPs) in response to multiple types of endogenous and exogenous stressors. SASP components are diverse in nature, including cytokines and chemokines, as well as a wide variety of soluble and insoluble factors, and may contribute to immune activation by promoting immune cell infiltration. While further research is definitely needed, it has recently been reported that islets from T1D mice, as well as β-cells from T1D donors, show elevated markers of senescence during disease progression [42], suggesting that β-cell senescence may be an adaptive response to long-term cellular stress, thereby potentially promoting autoimmunity through SASP [43].
Chronic inflammation and immune status in obesity
Concomitant obesity, diabetes, and cardiopathy suggest common molecular mechanisms for these diseases and new therapeutic targets. Cardiovascular diseases are considered the main cause of death and disability in diabetic patients. Diabetes exacerbates the pathologies underlying atherosclerosis and heart failure. Unfortunately, these mechanisms are not properly modulated by therapeutic strategies solely focused on optimal glycemic control with currently available drugs or approaches [44-47]. For example, glucose transporters may be attractive targets for therapy. Results of metabolomic, epigenetic, and transcriptomic analyses suggest that GLUT3 is related to glucose oxidation in mitochondria and ACLY-dependent acetyl-CoA generation, moreover, it is a rate-limiting step in the epigenetic regulation of inflammatory gene expression. Furthermore, inhibiting GLUT3-dependent acetyl-CoA generation is a promising way to mitigate various Th17-cell-mediated inflammatory diseases [48]. Furthermore, the activity of sodium–glucose linked transporters (SGLT) was shown to be 3-fold increased in the membrane vesicles in the intestine of diabetic subjects, which may increase capacity of glucose absorption in diabetic subjects [49]. Expression of SGLT1 in heart sarcolemma, which is mediated through leptin, was found to be upregulated in Type 2 diabetes mellitus (T2DM) and ischemia and decreased in Type 1 DM (T1DM). These SGLT1 levels were found to be correlated to insulin levels. Insulin stimulates SGLT1 activity mediated through protein kinase C activation, which in turn leads to the recruitment of SGLT1 to the plasma membrane. Increased levels of SGLT1 lead to increased uptake of glucose in cardiomyocytes of T2DM patients [50]. In obesity, an increase in inflammatory markers and mediators was shown to occur, along with an increase in metabolic syndrome components [49], and this phenomenon may be a precursor to arterial hypertension [51], type 2 diabetes [52], and cardiovascular events [53]. Highly sensitive C-reactive protein, an inflammatory marker, was shown to provide additional prognostic information for cardiovascular disease beyond traditional risk factors in all major patient populations [53, 54].
In diabetes, glucose is retained in kidney and gluconeogenesis is increased (partially via fructose-dependent pathway). This is not good for adaptation, because it maintains hyperglycemia. In many organisms including humans, the uptake and metabolism of D-glucose in cells constitutes a significant energy source [55, 56-54]. The brain alone, for its normal function, requires uptake of about 125 grams of glucose per day. Therefore, to provide a constant glucose delivery, blood glucose level is tightly controlled. This control involves hormones such as glucagon and insulin that regulate the glucose uptake into cells as well as its storage and endogenous production [57]. C-reactive protein (CRP) is an evolutionarily conserved protein [58]. From arthropods to humans, CRP has been found in every organism where the presence of CRP has been sought. Human CRP is a pentamer made up of five identical subunits which binds to phosphocholine (PCh) in a Ca2+-dependent manner.
Hyperlipidemia in atherosclerotic plaques leads to the recruitment and migration of monocytes and other immune and inflammatory cells into the subendothelial vascular layer. Any atherosclerotic lesions were shown to contain T cells in addition to macrophages, but T cells in humans with diabetes were found to have a predominantly proinflammatory Th1 phenotype [59, 60]. In addition to the detrimental effects of low-density lipoproteins on macrophages and foam cells, cholesterol crystals themselves in an atherosclerotic lesion can activate the protein-3 inflammatory complex [57, 58]. This leads to increased transcription of NF-κB-regulated gene products and interleukin-1β, providing an additional positive feedback mechanism to enhance the deleterious effects of cholesterol particles that accumulate in lipid-rich plaques [59, 60], which may be involved in accelerated atherosclerosis in diabetic dyslipidemia.
C-reactive protein (CRP) is an evolutionarily conserved protein, found in all species, where its presence was analyzed [61-63]. Human CRP is a pentamer made up of five identical subunits which may bind to phosphocholine in a manner that depends on Ca2+-. Increased CRP level is observed in humans with excessive body weight, since their adipocytes produce tumor necrosis factor α and interleukin 6, which are pivotal factors for CRP production stimulation. Furthermore, in healthy people hepatocytes produce relatively low rates of CRP compared to T2D patients, where elevated levels of inflammatory markers are reported, including CRP. CRP is also involved in dysfunctions of epithelium, the production of vasodilators, and vascular remodeling. Furthermore, increased CRP level is closely related to vascular system pathologies and metabolic syndrome [63]. CRP may be one of the possible targets for T2D therapy, and understanding the relationships between insulin and inflammation may be helpful for treatment and prevention of T2D. Along with that, ubiquitin ligases also may be involved in the control of the magnitude or duration of processes that govern outcome of cellular stress response. So, we may postulate a crosstalk among the fundamental processes governing ERS responses. In addition, an increased level of CRP may be a reliable predictor of vascular complications and progression of cardiovascular complications in patients with diabetes.
The pathology of diabetic cardiomyopathy remains poorly understood [65, 66]. Animal models of diabetes and studies based on cardiac imaging in humans have demonstrated both diastolic and systolic dysfunction. Diastolic dysfunction was shown to be a hallmark of diabetic cardiomyopathy, while systolic dysfunction represented the end stage of progressive disease [67, 68-]. Human studies also demonstrated early impairment of diastolic function, including decreased peak systolic and early diastolic myocardial rates [69].
Search for therapeutic approaches for the treatment of diabetes mellitus
The incidence of diabetes mellitus is increasing worldwide, and as recent studies have shown, it affects 7% of the global adult population [51]. There are two main types of diabetes mellitus: T1D, which is associated with complete or near-complete insulin deficiency due to autoimmune-mediated destruction of pancreatic β-cells; and T2D, which is associated with varying degrees of insulin resistance, impaired insulin secretion, moderate and severe β-cell apoptosis and increased hepatic glucose production [70].
It has been established that among the methods that reduce the pathological effect of T1D, the most efficient are continuous injections of insulin or transplantation of healthy pancreatic islets [71]. Complications that have traditionally been associated with diabetes mellitus include macrovascular conditions such as coronary heart disease, stroke and peripheral arterial disease, as well as microvascular conditions, including diabetic kidney disease, retinopathy and peripheral neuropathy [72]. Heart failure is also a common initial manifestation of cardiovascular disease in patients with type 2 diabetes mellitus [73] and is associated with a high risk of mortality in patients with T1D and T2D. Although a large disease burden associated with these traditional complications of diabetes still exists, the prevalence of these conditions is declining with improvements in type 2 diabetes therapies.
Epidemiological, animal, and in vitro studies support the antidiabetic effects of many dietary flavonoids [74, 75]. Dietary flavonoids exert their antidiabetic effects by acting on various cellular signaling pathways in the pancreas, liver, and skeletal muscle [76, 77]. Flavonoids were shown to be effective in relation to β-cell mass and function, as well as energy metabolism and insulin sensitivity in peripheral tissues. Many of the flavonoid-related studies discussed in this review also focus on specific signaling pathways involved in the effects of flavonoids on glucose homeostasis. Emerging evidence indicates that some dietary flavonoid metabolites act through several components of signaling cascades to modulate various cell types [78, 79]. However, studies on the antidiabetic effects of dietary flavonoid metabolites are scarce, and therefore, it is currently unknown whether certain dietary flavonoid metabolites may mediate the various biological actions of their parent molecules.
It is important to note that there are also some successes in the development of therapies against type 1 diabetes. Evidence indicates that defective mitochondrial function may play an important role in pancreatic β-cell dysfunction and apoptosis; however, the fundamental role of mitochondrial complex I in T1D and in β-cells remains unclear [80]. According to recent data, pancreatic cell protection may be achieved using the complex I inhibitor rotenone (ROT), and the potential mechanism was evaluated in a mouse model of T1D induced by streptozotocin (STZ) and in a cultured mouse pancreatic β-cell line Min6. ROT treatment had a hypoglycemic effect, restored insulin levels, and reduced inflammation and cell apoptosis in the pancreas. In vitro experiments also showed that ROT reduced β-cell apoptosis induced by STZ and inflammatory cytokines [81, 82]. In addition, there are data on the role of hormones, in particular estrogens, in the development of the physiopathology of pancreatic islets [83].
Moreover, consideration should be given to repurposing T2D drugs for the treatment of T1D to improve blood glucose control in recurrent metabolic and oxidative beta-cell stress and limit further immune degradation. These conceptually attractive new approaches will also require further support from in vitro molecular studies in primary human beta cells/islets and/or relevant models of beta cell lines.
As people with diabetes are now living longer, they become susceptible to a different set of complications (Fig. 1). Population studies show that vascular disease no longer accounts for the majority of deaths among people with diabetes, as it once did [76].
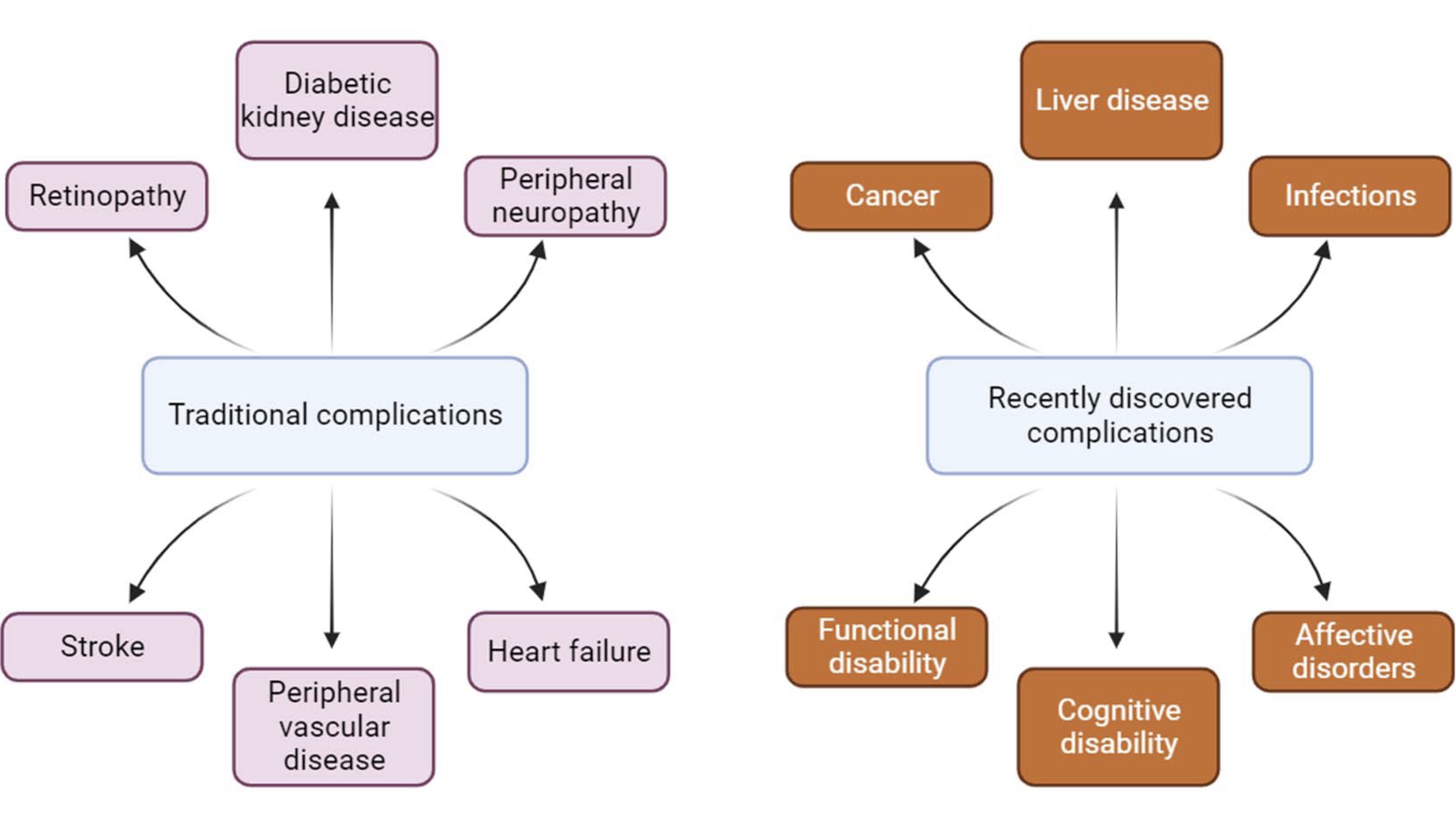
Fig. 1: Changes in diabetes complications in recent times.
Patients with T2D have been described as having epigenetic modifications. Epigenetics is the post-transcriptional modification of DNA or associated factors containing genetic information. These environmentally-influenced modifications, maintained during cell division, cause stable changes in gene expression. It was found the drug (apabetalone) is being studied to prevent major adverse cardiovascular events in people with T2D, low HDL cholesterol, chronic kidney failure, and recent coronary events. It was published a review aims to describe the relationship between obesity, long-term complications such as T2D, and epigenetic modifications and their possible treatments [77].
Cancer is now the leading cause of death for people with diabetes in some countries or regions [78-81], and the proportion of deaths from dementia has increased since the turn of the century. In England, traditional complications accounted for more than 50% of hospital admissions for people with diabetes in 2003 but only 30% in 2018, highlighting the changing nature of diabetes complications in recent times [79, 80].
It was demonstrated that autophagy promotes β-cell survival by delaying apoptosis and providing adaptive responses to mitigate the detrimental effects of ER stress and DNA damage [80-82], the latter of which is directly related to oxidative stress. Thus, elucidating the regulatory mechanisms of β-cell autophagy under diabetic conditions is an important area of research to better understand β-cell survival and develop therapies that target β-cells directly. Watada and Fujitani [83] comprehensively reviewed β-cell autophagy in the context of T2D in 2015. The following year, the Nobel Prize in Physiology or Medicine was awarded to Yoshinori Ohsumi for his "discoveries of the mechanisms of autophagy" [84], sparking further enthusiasm for how this fundamental cellular process might play a role in the development of diabetes. Overall, an autophagy-targeted approach to diabetes therapy requires caution and thoughtful design. An important consideration for developing therapies for diabetes, which is often administered chronically over a long period of time, is that autophagy has been reported to promote cancer cell survival [85], and inhibition of autophagy is of great clinical interest for the treatment of several cancers [86]. Epigenetics played a relevant role in prevention, diagnosis, and treatment of diabetes [87, 88].
Postoperative infection is also an important complication of diabetes. A meta-analysis revealed that diabetes mellitus was associated with surgical site infections [87]. Effect severity was greatest after cardiac procedures, and one study of patients in the U.S. undergoing coronary artery bypass grafting found that diabetes mellitus was an independent predictor of surgical site infection compared with nondiabetic patients [89-90].
Although diabetes mellitus does not appear to increase the risk of COVID-19 [91], various population-based studies report an increased risk of COVID-19 complications among patients with diabetes. In a population study in Scotland, patients with diabetes were found to have an increased risk of death or hospitalization in the intensive care unit for COVID-19 in comparison to subjects without diabetes [92]. The risk was especially high for patients with T1D. Both T1D and T2D were associated with a more than twofold increase in the risk of hospitalization with COVID-19 in a large-scale Swedish cohort study [93].
Conclusion
The incidence of diabetes mellitus is increasing worldwide; it has recently been reported that this disease affects 7% of adults worldwide. Diabetes mellitus is a complex metabolic disorder resulting from defects in insulin secretion. There are two main types of diabetes mellitus: T1D, which is associated with complete or near-complete insulin deficiency due to autoimmune-mediated destruction of pancreatic β-cells; and T2D, which is associated with varying degrees of insulin resistance, impaired insulin secretion, moderate and severe β-cell apoptosis and increased glucose production in the liver.
In this review, we have focused on the common features of T1D and T2D, which are islet-specific inflammatory microenvironments characterized by elevated local concentrations of proinflammatory cytokines, increased recruitment and activation of tissue innate and adaptive immune cells (e.g., macrophages, B cells and T cells) and the accumulation of amyloid deposits. As people with diabetes are now living longer, they become susceptible to a different set of complications. Population-based studies show that vascular disease no longer accounts for the majority of deaths among people with diabetes, as it once did. Cancer is now the leading cause of death for people with diabetes in some countries or regions, and the proportion of deaths from dementia has increased since the turn of the century. In England, traditional complications accounted for more than 50% of hospital admissions for people with diabetes in 2003 but only 30% in 2018, highlighting the changing nature of diabetes complications.
Acknowledgements
Authors thank AJE for help with language editing.
Funding
The authors gratefully acknowledge financial support from the Russian Scientific Foundation (grant no 23-24-00041).
Author contributions
All authors contributed to the article and approved the submitted version.
Disclosure Statement
The authors declare that the article was prepared in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
| 1 | Desai S, Deshmukh A. Mapping of type 1 diabetes mellitus. Curr Diabetes Rev 2020;16:438-441.
https://doi.org/10.2174/1573399815666191004112647 |
| 2 | Ilonen J, Lempainen J, Veijola R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat Rev Endocrinol 2019;15:635-650.
https://doi.org/10.1038/s41574-019-0254-y |
| 3 | Yahaya TO, Salisu TF. A review of type 2 diabetes mellitus predisposing genes. Curr Diabetes Rev 2019;16:52-61.
https://doi.org/10.2174/1573399815666181204145806 |
| 4 | Lisco G, Giagulli VA, De Pergola G, Guastamacchia E, Jirillo E, Triggiani V. Pancreatic macrophages and their diabetogenic effects: highlight on several metabolic scenarios and dietary approach. Endocr Metab Immune Disord Drug Targets 2023;23:304-315.
https://doi.org/10.2174/1871530322666220510123913 |
| 5 | Bowden SA. Partial remission (honeymoon phase) in type 1 diabetes mellitus. In: Atta-ur-Rahman (ed).: Frontiers in Clinical Drug Research. Diabetes and Obesity. Bentham Science Publishers; 2017 pp. 1-20.
https://doi.org/10.2174/9781681089348121070001 |
| 6 | Zhang L, Thurber GM.Imaging in diabete. In: Lewis JS, Keshari KR (eds): Imaging and metabolism. Cham, Switzerland: Springer International Publishing; 2018 pp. 175-197.
https://doi.org/10.1007/978-3-319-61401-4_8 |
| 7 | Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of japanese type II diabetic patients. Diabetologia 2002;45:85-96.
https://doi.org/10.1007/s125-002-8248-z |
| 8 | Lenzen S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic beta-cells. Biochim Biophys Acta 2017;1861:1929-1942.
https://doi.org/10.1016/j.bbagen.2017.05.013 |
| 9 | Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 2008;36:343-347.
https://doi.org/10.1042/BST0360343 |
| 10 | Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med 2014;66:75-87.
https://doi.org/10.1016/j.freeradbiomed.2013.07.036 |
| 11 | Bast A, Wolf G, Oberbaumer I, Walther R. Oxidative and nitrosative stress induces peroxiredoxins in pancreatic beta cells. Diabetologia 2002;45:867-76.
https://doi.org/10.1007/s00125-002-0846-1 |
| 12 | Novoselova EG, Glushkova OV, Parfenuyk SB, Khrenov MO, Lunin SM, Novoselova TV, et al.: Protective effect of peroxiredoxin 6 against toxic effects of glucose and cytokines in pancreatic RIN-m5F β-cells. Biochemistry (Moscow) 2019;84:637-643.
https://doi.org/10.1134/S0006297919060063 |
| 13 | Novoselova EG, Glushkova OV, Lunin SM, Khrenov MO, Parfenyuk SB, Novoselova TV, et al.: Peroxiredoxin 6 attenuates alloxan-induced type 1 diabetes mellitus in mice and cytokine-induced cytotoxicity in RIN-m5F Beta cells. J Diabetes Res 2020;2020:11.
https://doi.org/10.1155/2020/7523892 |
| 14 | Novoselova EG, Glushkova OV, Lunin SM., Khrenov MO, Parfenyuk SB, Novoselova TV, et al.: Thymulin and peroxiredoxin 6 have protective effects against streptozotocin induced type 1 diabetes in mice. Int J Immunopathol Pharmacol 2021;35:1-10.
https://doi.org/10.1177/20587384211005645 |
| 15 | Novoselova EG, Glushkova OV, Khrenov MO, Lunin SM, Novoselova TV, Parfenuyk SB. Role of innate immunity and oxidative stress in the development of type 1 diabetes mellitus. Peroxiredoxin 6 as a new antidiabetic agent. Biochemistry (Moscow) 2021;86:1579-1589.
https://doi.org/10.1134/S0006297921120075 |
| 16 | Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 1985;4:110-125.
https://doi.org/10.1159/000156969 |
| 17 | Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005;54(Suppl.S2):S97-S107.
https://doi.org/10.2337/diabetes.54.suppl_2.S97 |
| 18 | Rehman A, Nourooz-Zadeh J, Moller W, Tritschler H, Pereira P, Halliwell B. Increased oxidative damage to all dna bases in patients with typ eII diabetes mellitus. FEBS Lett 1999;448:120-122.
https://doi.org/10.1016/S0014-5793(99)00339-7 |
| 19 | Shin CS, Moon BS, Park KS, Kim SY, Park SJ, Chung MH, et al.: Serum 8-hydroxy-guanine levels are increased in diabetic patients. Diabetes Care 2001;24:733-737.
https://doi.org/10.2337/diacare.24.4.733 |
| 20 | National Diabetes Statistics Report. Estimates of Diabetes and Its Burden in the United States. Atlanta, GA: Centers for Disease Control and Prevention; 2014.
|
| 21 | Groop LC, Bottazzo GF, Doniach D. Islet cell antibodies identify latent type i diabetes in patients aged 35-75 years at diagnosis. Diabetes 1986;35:237-241.
https://doi.org/10.2337/diab.35.2.237 |
| 22 | Tuomi T, Groop LC, Zimmet PZ, Rowley MJ, Knowles W, Mackay IR. Antibodies to glutamic acid decarboxylase reveal latent autoimmune diabetes mellitus in adults with a non-insulin-dependent onset of disease. Diabetes 1993;42:359-362.
https://doi.org/10.2337/diab.42.2.359 |
| 23 | Nourooz-Zadeh J, Tajaddini-Sarmadi J, McCarthy S, Betteridge DJ, Wolff SP. Elevated levels of authentic plasma hydroperoxides in NIDDM. Diabetes 1995;44:1054-1058.
https://doi.org/10.2337/diab.44.9.1054 |
| 24 | Elizabeth MM, Alarcon-Aguilar JF, Clara OC, Escobar-Villanueva, Del Carmen M.. Pancreatic β-cells and type 2 diabetes development. Curr Diabetes Rev 2017;13:108-121.
https://doi.org/10.2174/1573399812666151020101222 |
| 25 | Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 2016; 3:79-94.
https://doi.org/10.1007/s00018-015-2052-6 |
| 26 | Ghiselli A, Laurenti O, De Mattia G, Maiani G, Ferro-Luzzi A. Salicylate hydroxylation as an early marker of in vivo oxidative stress in diabetic patients. Free Radic Biol Med 1992;13:621-626.
https://doi.org/10.1016/0891-5849(92)90036-G |
| 27 | Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 2016;73:79-94.
https://doi.org/10.1007/s00018-015-2052-6 |
| 28 | Chen J, Stimpson SE, Fernandez-Bueno GA, Mathews CE. Mitochondrial reactive oxygen species and type 1 diabetes. Antioxid Redox Signal 2018;29:1361-1372.
https://doi.org/10.1089/ars.2017.7346 |
| 29 | Las G, Oliveira MF, Shirihai OS. Emerging roles of beta-cell mitochondria in type-2-diabetes. Mol Aspects Med 2020;71:100843.
https://doi.org/10.1016/j.mam.2019.100843 |
| 30 | Qureshi MA, Haynes CM, Pellegrino MW. The mitochondrial unfolded protein response: Signaling from the powerhouse. J Biol Chem 2017;292:13500-13506.
https://doi.org/10.1074/jbc.R117.791061 |
| 31 | Naresh NU, Haynes CM. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb Perspect Biol 2019;11:1-17.
https://doi.org/10.1101/cshperspect.a033944 |
| 32 | Detaille D, Guigas B, Chauvin C, Batandier C, Fontaine E, Wiernsperger N, et al.: Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 2005;54:2179-2187.
https://doi.org/10.2337/diabetes.54.7.2179 |
| 33 | Guigas B, Detaille D, Chauvin C, Batandier C, De Oliveira F, Fontaine E, et al.: Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem J. 2004;382:877-884.
https://doi.org/10.1042/BJ20040885 |
| 34 | Beery ML, Jacobsen LM, Atkinson MA, Butler AE, Campbell-Thompson M. Islet amyloidosis in a child with type 1 diabetes. Islets 2019;11:44-49.
https://doi.org/10.1080/19382014.2019.1599707 |
| 35 | Westermark GT, Krogvold L, Dahl-Jorgensen K, Ludvigsson J. Islet amyloid in recent-onset type 1 diabetes-the DiViD study. Ups J Med Sci 2017;122:201-203.
https://doi.org/10.1080/03009734.2017.1359219 |
| 36 | Ghiasi SM, Dahllof MS, Osmai Y, Osmai M, Jakobsen KK, Aivazidis A, et al.: Regulation of the beta-cell inflammasome and contribution to stress-induced cellular dysfunction and apoptosis. Mol Cell Endocrinol 2018;478:106-114.
https://doi.org/10.1016/j.mce.2018.08.001 |
| 37 | Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al.: Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 2011;17:1481-1489.
https://doi.org/10.1038/nm.2513 |
| 38 | Zhou T, Hu Z, Yang S, Sun L, Yu Z, Wang G. Role of adaptive and innate immunity in type 2 diabetes mellitus. J Diabetes Res 2018;2018:7457269.
https://doi.org/10.1155/2018/7457269 |
| 39 | Batandier C, Guigas B, Detaille D, El-Mir MY, Fontaine E, Rigoulet M, et al.: The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr 2006;38:33-42.
https://doi.org/10.1007/s10863-006-9003-8 |
| 40 | Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab 2019;29:1045-1060.e10.
https://doi.org/10.1016/j.cmet.2019.01.021 |
| 41 | Li N, Liu F, Yang P, Xiong F, Yu Q, Li J, et al.: Aging and stress induced beta cell senescence and its implication in diabetes development. Aging (Albany NY) 2019;11:9947-9959.
https://doi.org/10.18632/aging.102432 |
| 42 | Kondo M, Tanabe K, Amo-Shiinoki K, Hatanaka M, Morii T, Takahashi H, et al.: Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia 2018;61:2189-2201.
https://doi.org/10.1007/s00125-018-4679-y |
| 43 | Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al.: Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 2016;23:303-314.
https://doi.org/10.1016/j.cmet.2015.11.011 |
| 44 | Fattah H, Vallon V. The Potential Role of SGLT2 Inhibitors in the treatment of type 1 diabetes mellitus. Drugs 2018;78:717-726.
https://doi.org/10.1007/s40265-018-0901-y |
| 45 | Asahara SI, Ogawa W. SGLT2 inhibitors and protection against pancreatic beta cell failure. Diabetol Int 2019;10:1-2.
https://doi.org/10.1007/s13340-018-0374-y |
| 46 | Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, et al.: Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat Med 2018;24:1108-1112.
https://doi.org/10.1038/s41591-018-0089-4 |
| 47 | Xu G, Chen J, Jing G, Shalev A. Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes 2012;61:848-856.
https://doi.org/10.2337/db11-0955 |
| 48 | Dyer J, Wood IS, Palejwala A, Ellis A, Shirazi-Beechey SP. Expression of monosaccharide transporters in intestine of diabetic humans. Am J Physiol Gastrointest Liver Physiol. 2002;282:G241-G248 Doi: 10.1152/ajpgi.00310.2001.
https://doi.org/10.1152/ajpgi.00310.2001 |
| 49 | Banerjee SK, McGaffin KR, Pastor-Soler NM, Ahmad F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res. 2009;84:111-118 doi: 10.1093/cvr/cvp190.
https://doi.org/10.1093/cvr/cvp190 |
| 50 | Maamoun H, Abdelsalam SS, Zeidan A, Korashy HM, Agouni A. Endoplasmic reticulum stress: a critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int J Mol Sci 2019; 20:e1658.
https://doi.org/10.3390/ijms20071658 |
| 51 | Aguayo-Mazzucato C. Functional changes in beta cells during ageing and senescence. Diabetologia 2020;63:2022-2029.
https://doi.org/10.1007/s00125-020-05185-6 |
| 52 | Low Wang CC, Hess CN, Hiatt WR, Goldfine AB. Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus - mechanisms, management, and clinical considerations. Circulation 2016;133:2459-2502.
https://doi.org/10.1161/CIRCULATIONAHA.116.022194 |
| 53 | Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA 1999;282:2131-2135.
https://doi.org/10.1001/jama.282.22.2131 |
| 54 | Shim K, Begum R, Yang C, Wang H. Complement activation in obesity, insulin resistance, and type 2 diabetes mellitus. World J Diabetes 2020;11:1-12.
https://doi.org/10.4239/wjd.v11.i1.1 |
| 55 | Esser N, Paquot N, Scheen AJ. Inflammatory markers and cardiometabolic diseases. Acta Clin Belg 2015;70:193-199.
https://doi.org/10.1179/2295333715Y.0000000004 |
| 56 | Kowalski GM, Bruce CR. The regulation of glucose metabolism: Implications and considerations for the assessment of glucose homeostasis in rodents. Am J Physiol Endocrinol Metab 2014;307:E859-E871.
https://doi.org/10.1152/ajpendo.00165.2014 |
| 57 | Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003;107:391-397.
https://doi.org/10.1161/01.CIR.0000055014.62083.05 |
| 58 | Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA 2003;290:2945-2951.
https://doi.org/10.1001/jama.290.22.2945 |
| 59 | Vozarova B, Weyer C, Lindsay RS, Pratley RE, Bogardus C, Tataranni PA. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002;51:455-461.
https://doi.org/10.2337/diabetes.51.2.455 |
| 60 | Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al.: NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357-1361.
https://doi.org/10.1038/nature08938 |
| 61 | Sun X, He S, Wara AK, Icli B, Shvartz E, Tesmenitsky Y, et al.: Systemic delivery of microRNA-181b inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ Res 2014;114:32-40.
https://doi.org/10.1161/CIRCRESAHA.113.302089 |
| 62 | Cuaz-Perolin C, Billiet L, Bauge E, Copin C, Scott-Algara D, Genze F, et al.: Antiinflammatory and antiatherogenic effects of the NF-kappaB inhibitor acetyl-11-keto-beta-boswellic acid in LPS-challenged ApoE−/− mice. Arterioscler Thromb Vasc Biol 2008;28:272-277.
https://doi.org/10.1161/ATVBAHA.107.155606 |
| 63 | Pathak A, Agrawal A. Evolution of C-reactive protein. Front Immunol 2019;10: 943.
https://doi.org/10.3389/fimmu.2019.00943 |
| 64 | Stanimirovic J, Radovanovic J, Banjac K, Obradovic M, Essack M, Zafirovic S,et al.: Role of C-reactive protein in diabetic inflammation.. Mediators Inflamm 2022;2022: 3706508.
https://doi.org/10.1155/2022/3706508 |
| 65 | Maisch B, Alter P, Pankuweit S. Diabetic cardiomyopathy--fact or fiction? Herz 2011;36:102-115.
https://doi.org/10.1007/s00059-011-3429-4 |
| 66 | Davies JL, Kawaguchi Y, Bennett ST, Copeman JB, Cordell HJ, Pritchard LE, et al.: A genome-wide search for human type 1 diabetes susceptibility genes. Nature 1994;371:130-136.
https://doi.org/10.1038/371130a0 |
| 67 | Borges GR, de Oliveira M, Salgado HC, Fazan R Jr. Myocardial performance in conscious streptozotocin diabetic rats. Cardiovasc Diabetol 2006;5:26.
https://doi.org/10.1186/1475-2840-5-26 |
| 68 | Dillmann WH. Diabetic Cardiomyopathy. Circ Res 2019;124:1160-1162.
https://doi.org/10.1161/CIRCRESAHA.118.314665 |
| 69 | Cole JB, Florez JC. Genetics of diabetes mellitus and diabetes complications. Nat Rev Nephrol 2020;16:377-390.
https://doi.org/10.1038/s41581-020-0278-5 |
| 70 | Shah AD, Langenberg C, Rapsomaniki E, Denaxas S, Pujades-Rodriguez M, Gale CP, et al.: Type 2 diabetes and incidence of cardiovascular diseases: a cohort study in 1•9 million people. Lancet Diabetes Endocrinol 2015;3:105-113.
https://doi.org/10.1016/S2213-8587(14)70219-0 |
| 71 | Naqvi RA, Naqvi AR, Singh A, Priyadarshini M, Balamurugan AN, Layden BT. The future treatment for type 1 diabetes: Pig islet- or stem cell-derived β cells? Front Endocrinol (Lausanne) 2023;13:1001041.
https://doi.org/10.3389/fendo.2022.1001041 |
| 72 | Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001;414:782-787.
https://doi.org/10.1038/414782a |
| 73 | Bertoni AG, Hundley WG, Massing MW, Bonds DE, Burke GL, Goff DC Jr. Heart failure prevalence, incidence, and mortality in the elderly with diabetes. Diabetes Care 2004;27:699-703.
https://doi.org/10.2337/diacare.27.3.699 |
| 74 | Babu PV, Liu D, Gilbert ER. Recent advances in understanding the anti-diabetic actions of dietary flavonoids. J Nutr Biochem 2013;24:1777-1789.
https://doi.org/10.1016/j.jnutbio.2013.06.003 |
| 75 | Crozier A, Jaganath IB, Clifford MN. Dietary phenolics: chemistry, bioavailability and effects on health. Nat Prod Rep 2009;26:1001-1043.
https://doi.org/10.1039/b802662a |
| 76 | Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, Williams DE, et al.: Changes in diabetes-related complications in the United States, 1990-2010 N Engl J Med 2014; 370:1514-1523.
https://doi.org/10.1056/NEJMoa1310799 |
| 77 | Philippe J, Raccah D. Treating type 2 diabetes: how safe are current therapeutic agents? Int J Clin Pract 2009;63:321-332.
https://doi.org/10.1111/j.1742-1241.2008.01980.x |
| 78 | Akkati S, Sam KG, Tungha G. Emergence of promising therapies in diabetes mellitus. J Clin Pharmacol 2011;51:796-804.
https://doi.org/10.1177/0091270010376972 |
| 79 | Pearson-Stuttard J, Bennett J, Cheng YJ, Vamos EP, Cross AJ, Ezzati M, et al.: Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: an epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol 2021;9:165-173.
https://doi.org/10.1016/S2213-8587(20)30431-9 |
| 80 | Pearson-Stuttard J, Buckley J, Cicek M, Gregg EW. The changing nature of mortality and morbidity in patients with diabetes. Endocrinol Metab Clin North Am 2021;50:357-368.
https://doi.org/10.1016/j.ecl.2021.05.001 |
| 81 | Pearson-Stuttard J, Cheng YJ, Bennett J, Vamos EP, Zhou B, Valabhji J, et al.: Trends in leading causes of hospitalisation of adults with diabetes in England from 2003 to 2018: an epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol 2022;10:46-57.
https://doi.org/10.1016/S2213-8587(21)00288-6 |
| 82 | Hayes HL, Peterson BS, Haldeman JM, Newgard CB, Hohmeier HE, Stephens SB. Delayed apoptosis allows islet β-cells to implement an autophagic mechanism to promote cell survival. PLoS One 2017;12:e0172567.
https://doi.org/10.1371/journal.pone.0172567 |
| 83 | Watada H, Fujitani Y. Minireview: autophagy in pancreatic β-cells and its implication in diabetes. Mol Endocrinol 2015;29:338-348.
https://doi.org/10.1210/me.2014-1367 |
| 84 | Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: breakthroughs in baker's yeast fuel advances in biomedical research. Proc Natl Acad Sci USA 2017;114:201-205.
https://doi.org/10.1073/pnas.1619876114 |
| 85 | White E. The role for autophagy in cancer. J Clin Invest 2015;125:42-46.
https://doi.org/10.1172/JCI73941 |
| 86 | Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 2017;16:487-511.
https://doi.org/10.1038/nrd.2017.22 |
| 87 | Ling C. Epigenetic regulation of insulin action and secretion - role in the pathogenesis of type 2 diabetes. J Intern Med 2020;288:158-167.
https://doi.org/10.1111/joim.13049 |
| 88 | Sommese L, Benincasa G, Lanza M, Sorriento A, Schiano C, Lucchese R, et al.: Novel epigenetic-sensitive clinical challenges both in type 1 and type 2 diabetes. J Diabetes Complications 2018;32:1076-1084.
https://doi.org/10.1016/j.jdiacomp.2018.08.012 |
| 89 | Martin ET, Kaye KS, Knott C, Nguyen H, Santarossa M, Evans R, et al.: Diabetes and risk of surgical site infection: a systematic review and meta-analysis. Infect Control Hosp Epidemiol 2016;37:88-99.
https://doi.org/10.1017/ice.2015.249 |
| 90 | Trussell J, Gerkin R, Coates B, Brandenberger J, Tibi P, Keuth J, et al.: Impact of a patient care pathway protocol on surgical site infection rates in cardiothoracic surgery patients. Am J Surg 2008;196:883-889.
https://doi.org/10.1016/j.amjsurg.2008.07.024 |
| 91 | Apicella M, Campopiano MC, Mantuano M, Mazoni L, Coppelli A, Del Prato S. COVID-19 in people with diabetes: understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol 2020;8:782-792.
https://doi.org/10.1016/S2213-8587(20)30238-2 |
| 92 | McGurnaghan SJ, Weir A, Bishop J, Kennedy S, Blackbourn LAK, McAllister DA, et al.: Risks of and risk factors for COVID-19 disease in people with diabetes: a cohort study of the total population of Scotland. Lancet Diabetes Endocrinol 2021;9:82-93.
https://doi.org/10.1016/S2213-8587(20)30405-8 |
| 93 | Rawshani A, Kjölhede EA, Rawshani A, Sattar N, Eeg-Olofsson K, Adiels M, et al. Severe COVID-19 in people with type 1 and type 2 diabetes in Sweden: A nationwide retrospective cohort study. Lancet Reg Health Eur 2021;4:100105.
https://doi.org/10.1016/j.lanepe.2021.100105 |